
纳米抗体(Nanobody)由于其结构简单、稳定性高以及易于工程化改造,近年来在抗体工程和生物医药研究中受到了广泛关注。与传统抗体相比,纳米抗体仅由驼科重链抗体的可变区(VHH)构成,分子量通常只有约15 kDa。这种单域结构不仅赋予纳米抗体更好的溶解性和稳定性,也使其更容易通过基因工程进行模块化设计。然而,单个纳米抗体在某些应用中仍然存在局限,例如结合能力有限、功能单一以及在复杂生物系统中难以产生足够强的生物学效应。因此,近年来研究重点逐渐转向纳米抗体的多聚化(multimerization)和多特异性(multispecificity)工程设计,通过组合多个纳米抗体模块来提升其功能性能。
纳米抗体多价化的核心思想是利用多个抗体结构域协同作用,从而增强整体结合能力和生物学功能。当多个纳米抗体同时作用于同一靶标时,会产生明显的“亲合力效应(avidity principle)”,使抗体整体亲合力显著高于单个结合位点。这种效应在抗病毒抗体、肿瘤靶向抗体以及受体调控研究中尤为重要。例如,在病毒中和研究中,多价纳米抗体可以同时结合病毒表面的多个表位,从而显著提高中和能力。在肿瘤免疫治疗领域,多价纳米抗体还可以通过诱导受体聚集来调控细胞信号通路,从而增强治疗效果。正因如此,如何高效构建多价纳米抗体成为抗体工程研究的重要方向。
目前纳米抗体多价化的技术路线大致可以分为两大类:体内组装策略(in vivo assembly)和体外工程化构建策略(in vitro engineering)。体内组装策略主要依赖细胞表达系统,通过基因工程设计,使多个纳米抗体在表达过程中自动形成稳定的多聚结构。这种策略的优势在于表达过程简单、结构均一性较高,并且适合大规模生产,因此在纳米抗体药物开发中得到了广泛应用。
最直接的纳米抗体多价化方法是通过柔性连接肽(linker)将多个VHH结构域在基因水平串联。在这种设计中,多个纳米抗体序列被克隆到同一表达载体中,并通过短肽序列连接形成单一多结构域蛋白。在表达系统中翻译后,该蛋白即形成多价纳米抗体结构。
最常用的linker是富含甘氨酸和丝氨酸残基的柔性肽序列,例如经典的(G4S)n linker。这种linker具有较高柔性,可以减少不同结构域之间的空间干扰,使每个纳米抗体结构域能够独立折叠并保持其抗原结合能力。通过调节linker的长度,可以在一定程度上控制纳米抗体结构域之间的空间距离,从而优化多价结合效果。
除了常用的柔性linker外,弹性蛋白样多肽(ELP)结构域也可作为结构支架,用于组织多个纳米抗体形成更复杂的多价结构。ELP通常由重复的五肽序列(VPGXG)n构成,具有良好的可塑性和结构稳定性,为纳米抗体多价化提供了一种更加灵活的结构框架。
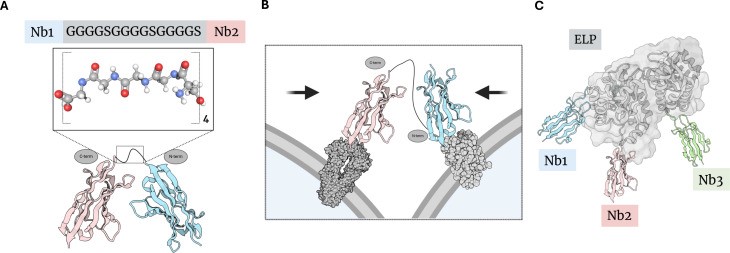
图1 柔性linker串联表达
(A)在纳米抗体Nb1和Nb2之间引入(Gly4Ser)3柔性linker,构建形成一个双价纳米抗体结构。
(B)通过(Gly4Ser)3 linker将两个不同的纳米抗体进行连接,构建形成双特异性纳米抗体结构。这种结构可以形成类似“分子夹钳(molecular clamp)”的构象,使表达相应抗原的两个细胞在空间上更加接近。
(C)ELP结构域可以用于容纳并连接三个不同的纳米抗体结构域,构建三特异性纳米抗体结构。
在抗病毒研究中,研究者通过linker将多个识别同一病毒表位的纳米抗体连接,形成多价结构,从而显著增强病毒中和活性。在肿瘤靶向应用中,也可以将识别不同肿瘤抗原或受体的VHH结构域串联,实现多位点结合或受体聚集,从而增强信号阻断或细胞定向效应。类似策略还被探索用于多特异性免疫调控,例如同时靶向肿瘤抗原和免疫检查点受体,以提高治疗选择性和效力。
另一种非常重要的体内多聚化策略是Fc融合表达(VHH-Fc fusion)。在这种设计中,纳米抗体的C端与IgG抗体的Fc结构域进行基因融合。由于Fc结构域天然能够形成稳定的二聚体结构,因此在表达过程中,两个VHH-Fc融合蛋白会通过Fc区域自发形成二聚体,从而实现纳米抗体的二价化。
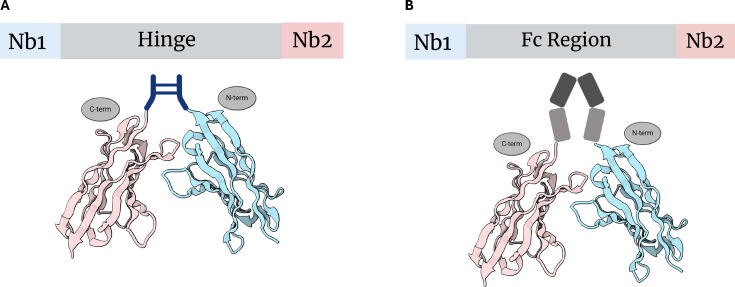
图2 Fc融合表达
(A)在纳米抗体Nb1与Nb2之间引入铰链(hinge)区序列,从而构建形成双价(bivalent)的多聚纳米抗体(polybody)结构。
(B)在纳米抗体Nb1与Nb2之间引入Fc区(crystallizable Fc region)序列,同样构建出双价(bivalent)多聚纳米抗体结构。
这种设计不仅能够增加纳米抗体的结合价数,还可以显著改善其药代动力学特性。传统单域纳米抗体由于分子量较小,在体内往往会被快速肾脏清除,而Fc融合可以增加分子量,并通过与FcRn受体结合延长循环半衰期。此外,Fc结构域还能够介导多种免疫效应功能,例如抗体依赖性细胞毒作用(ADCC)和补体依赖性细胞毒作用(CDC),从而在免疫治疗中发挥更强的生物学效应。因此,VHH-Fc融合蛋白已经成为当前纳米抗体药物开发中最常见的分子形式之一,在肿瘤治疗和抗病毒抗体开发中均有广泛应用。
除了linker串联和Fc融合之外,研究人员还利用多种自组装卷曲螺旋肽(self‑assembling coiled coils and peptides)序列来驱动纳米抗体的多聚化。这些结构域能够在表达过程中通过特定蛋白—蛋白相互作用自发形成稳定的寡聚体,从而将多个纳米抗体模块组装在一起,实现多价或多特异性构建。
例如来自Staphylothermus marinus的右手卷曲螺旋(RHCC)能够形成四聚体结构,将纳米抗体融合到RHCC后可自发生成四价复合物,从而增强整体结合力。类似策略也包括COMPcc,一种由软骨寡聚基质糖蛋白衍生的五链卷曲螺旋结构域,其融合纳米抗体后可形成五价复合物。
此外,这类自组装策略不仅可以在细胞内表达时自发形成,还可以与SpyTag–SpyCatcher系统等模块化共价连接方法结合,在体外精确组装不同数量的纳米抗体模块。利用这种策略,可以将多个纳米抗体模块按需拼接,构建多价或多特异性纳米抗体结构,在不影响单个抗体抗原识别能力的前提下显著增强多价结合效应和功能活性。
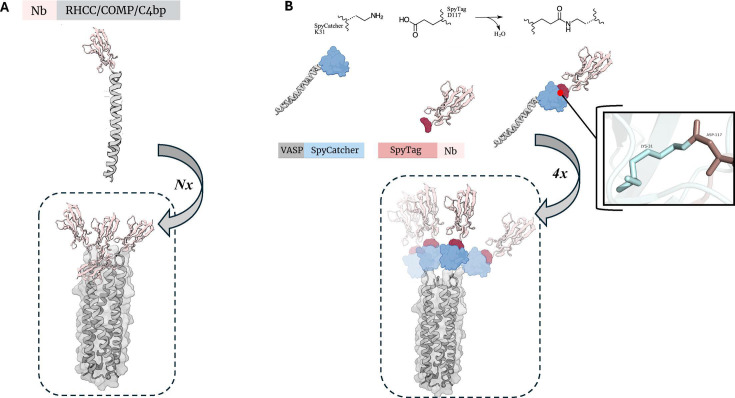
图3 自组装卷曲螺旋肽接导的多聚化
(A)编码自组装coiled-coil结构域的序列被连接到Nb编码序列上。所得的Nb-coiled-coil融合蛋白能够与其他相同的融合蛋白自组装。
(B)利用SpyTag-SpyCatcher系统,将Nb与血管扩张刺激磷蛋白(VASP)进行连接,从而形成一种四聚体结构。
除了利用卷曲螺旋结构域形成有限数目的寡聚体外,研究人员还开发了利用天然蛋白纳米颗粒(protein nanoparticles)作为支架来展示多个纳米抗体结构域的策略。这类方法通常依赖能够自发组装成高阶对称结构的蛋白质,例如细菌毒素亚基、铁蛋白或其他多亚基酶复合体。通过将纳米抗体与这些蛋白亚基进行基因融合,可以在表达过程中形成高度有序的多价抗体结构。
与依赖基因表达系统的体内组装策略不同,体外工程化方法通过酶促或化学介导在蛋白翻译后阶段实现纳米抗体的定向连接。这类策略具有高度灵活性,可以在不同纳米抗体模块之间实现可控拼接,并允许在保持各结构域功能完整的情况下构建复杂的多特异性结构。
一种常见的酶促方法是利用Sortase A转肽酶进行定点连接。Sortase A能够识别特定的LPXTG序列,并在该位点形成酰基中间体,随后通过甘氨酸残基的亲核攻击完成蛋白之间的共价连接。通过在纳米抗体分子末端引入Sortase识别序列,可以实现两个或多个纳米抗体之间的精确定向连接,从而构建双价或多价结构。这种方法具有较高的位点特异性,因此能够避免随机化学偶联带来的结构异质性问题。
除了酶促连接外,化学介导方法也被广泛应用于纳米抗体多聚化。例如,异型双功能交联剂Sulfo-SMCC可以同时与蛋白的氨基和巯基反应,从而形成稳定的硫醚键。通过在纳米抗体上引入特定的反应基团,可以实现不同纳米抗体之间的定向交联。虽然化学介导方法在结构控制方面不如酶促精确,但其操作简单、适用范围广,因此仍然是实验室和工业生产中常用的技术路线。
除了传统的共价连接方法,研究人员还提出了一种利用膜蛋白聚集特性的纳米抗体组装策略。这种方法通过在纳米抗体C端引入单跨膜结构域,使其在表达过程中定位于脂质膜环境中。由于跨膜结构域具有明显的疏水性,这些融合蛋白会倾向于互相聚集,将其跨膜区域隐藏于疏水环境(如膜或膜模拟剂)中,从而促进纳米抗体之间的空间靠近。
为了在体外维持这种结构稳定性,研究人员利用一种称为peptidisc的膜模拟体系对跨膜结构域进行包裹。Peptidisc是一种两亲性多肽结构,其内部疏水区域可以稳定跨膜片段,而外部亲水区域则保证整体复合物在水溶液中的溶解性。通过这种方式,可以形成包含多个纳米抗体的多价复合物。
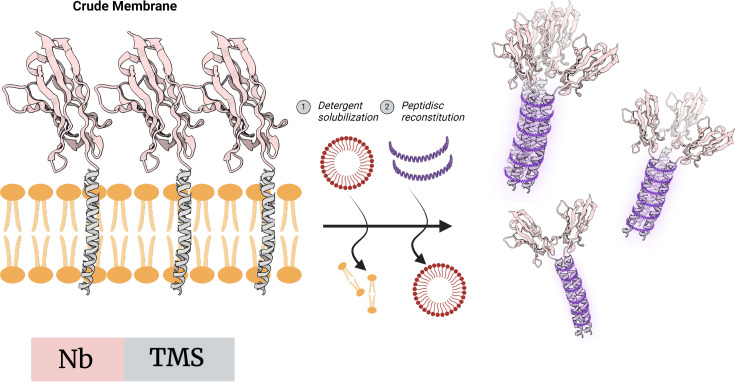
图4 TMS–Nb嵌合体在膜模拟体系中聚集
实验结果表明,这种膜模拟驱动的纳米抗体聚集结构能够显著提高抗原结合能力。例如,在抗GFP纳米抗体系统中,多价聚集结构表现出明显的亲合力增强效应。此外,在ELISA检测体系中,这种多价纳米抗体复合物还能够显著提高检测灵敏度,最高可提升30倍以上。尽管该方法在价数控制方面仍存在一定挑战,但其通过物理聚集驱动多价化的设计思路,为纳米抗体工程提供了一种新的技术方向。
随着多价和多特异性纳米抗体工程技术的不断发展,这类分子在多个生物医药领域展现出广阔的应用潜力。首先,在抗病毒研究中,多价纳米抗体能够同时结合病毒表面的多个表位,从而显著提高中和效率,并降低病毒逃逸突变带来的风险。其次,在肿瘤免疫治疗领域,多特异性纳米抗体可以同时靶向肿瘤抗原和免疫细胞受体,从而实现类似双特异性抗体或T细胞连接器的功能。此外,在诊断和生物分析领域,多价纳米抗体还可以提高检测灵敏度。例如,在免疫检测或生物传感器系统中,通过提高抗体价数可以增强抗原捕获能力,从而显著降低检测限。这一特点使纳米抗体多价化技术在病毒检测、肿瘤标志物检测以及环境监测等领域具有重要价值。
总体而言,纳米抗体多价化与多特异性工程已经成为抗体工程领域的重要研究方向。通过结合体内表达系统、自组装蛋白结构以及体外酶促连接等多种技术路线,研究人员能够根据不同应用需求构建结构多样、功能丰富的纳米抗体分子。这些技术不仅推动了新一代抗体药物的开发,也为诊断试剂和生物分析工具的设计提供了新的思路。
参考文献
Al-Seragi M, Chen Y, Duong van Hoa F. Advances in nanobody multimerization and multispecificity: from in vivo assembly to in vitro production. Biochem Soc Trans. 2025 Feb 7;53(1):235–48. doi: 10.1042/BST20241419.
COVID-19将中和抗体(neutralizing antibody, nAb)的概念引入了公众视野,并提升了这类抗体在科学界的整体关注度。与许多病毒情况类似,血清中和抗体水平(通过体外试验测定)与对SARS-CoV-2的感染和疾病保护作用之间存在相当好的相关性。然而,对于中和抗体的作用方式仍存在一些误解。‘中和’一词常被理解为抗体仅通过阻断感染发挥功能。此外,中和抗体的作用机制也存在巨大差异。
关于中和作用的定义,目前主要有两种被广泛接受的观点:
上述两种定义下,中和作用都可以通过体外实验检测。然而,“中和”一词在某些场景中也被用于描述抗体在体内的抗病毒活性,这并不总是等同于狭义上的阻断病毒进入。因此,绝大多数研究在提及中和抗体时,会明确限定其定义。
中和抗体能够阻断有包膜病毒进入靶细胞,其前提是抗体必须与病毒颗粒表面的功能性进入分子,通常是包膜(Env)蛋白的刺突结构结合。与这些功能性结构结合,不仅赋予中和抗体阻止病毒进入的能力,还可能在体内触发多种其他抗病毒效应,而这些效应通常在体外中和实验中无法体现。例如,抗体与病毒颗粒结合后,可以通过抗体的Fc片段与Fc受体(FcRs)相互作用,从而被吞噬细胞摄取,或被其他携带FcR的细胞捕获,进而阻止病毒接触靶细胞。抗体识别的功能性病毒结构也可能在受感染细胞表面表达,使这些细胞成为抗体依赖性细胞介导的细胞毒作用(ADCC)的靶标,并增强抗体抑制细胞间病毒传播的能力。
除了中和抗体外,一些非中和抗体(non-neutralizing antibody, nnAb)也能够通过Fc依赖的方式发挥抗病毒活性。这些作用既可以在包含效应细胞的体外实验中观察到,也能在体内的预防或治疗感染环境中显现。非中和抗体的作用机制可以分为以下几类:
在实际感染过程中,病毒颗粒的部分功能性分子可能发生解体或构象变化,从而暴露出平时不可及的表位。非中和抗体便可以识别这些表位,并通过补体激活、吞噬作用等途径触发免疫效应。类似的机制同样适用于非中和抗体对受感染细胞的作用。然而,总体来看,中和抗体在体内的抗病毒效果通常显著优于非中和抗体,这也解释了为何研究更聚焦于中和抗体。
与病毒体上的天然结构具有足够亲和力结合的抗体都有可能干扰病毒感染靶细胞。由抗体介导的体外中和过程可通过多种不同机制实现,以下将讨论体外病毒中和的主要机制,尤其针对包膜病毒。
中和抗体可通过与病毒表面包膜蛋白结合,直接干扰病毒与宿主受体的相互作用,从而阻断病毒感染。这一机制可归纳为“占位(occupancy)或涂层(coating)模型”:当抗体占据病毒颗粒表面足够比例的可接近表位时,会在空间上阻止病毒附着受体,或抑制膜融合等进入步骤。该模型强调抗体的中和能力与其对病毒表面抗原的亲和力密切相关。抗体的亲和力越强,占据表位越为稳定,中和效果越好。结构学研究进一步揭示,抗体多样化机制可以使其识别病毒表面广泛的抗原表位,锁定病毒包膜中的脆弱位点,通过多种结合策略干扰或破坏刺突蛋白的结构稳定性。如许多包膜蛋白(如Env分子)以亚稳态存在于病毒颗粒的预融合构象中,与宿主受体结合后需经历显著的构象重排以介导膜融合,促使细胞感染。多项研究显示,中和抗体与重组Env蛋白结合可诱导其构象改变,使其失去功能,从而不可逆地阻断病毒进入靶细胞。
抗体分子具有较大的尺寸,与典型病毒刺突蛋白相当。空间阻断的核心机制在于中和抗体通过物理占位阻止病毒与靶细胞受体的结合。这一过程依赖抗体与病毒受体结合蛋白的相互作用,尤其是直接参与受体结合的关键表位。抗体的中和效应与其亲和力密切相关,亲和力越高,对受体结合的竞争抑制越显著。
抗体介导的病毒颗粒聚集通常被视为区别于直接阻断感染的中和机制,但由于该过程可显著降低病毒的感染力,因此可归类为一种特殊形式的中和作用。其发生动力学通常呈现抗体浓度依赖性的钟形曲线:在低抗体水平下,抗体在单个病毒颗粒表面实现高效交联,从而促进颗粒间的初始聚集;随着抗体浓度升高,抗体通过桥接作用进一步驱动不同病毒颗粒间的聚合;然而,当病毒表面的表位逐渐被抗体饱和后,交联机会减少,聚集水平随之下降。
在体内环境中,聚集效应的显著性取决于多种因素,包括靶细胞的可及性、体液中抗体与病毒颗粒的比例以及免疫系统背景。值得注意的是,免疫细胞能够识别并吞噬这些由抗体介导形成的聚集体,不仅通过直接清除病毒实现中和,还可能触发补体活化、Fc 受体依赖性吞噬等间接抗病毒机制。这一过程凸显了抗体–病毒相互作用的复杂性,并在一定程度上解释了体内抗体应答的多样性和功能拓展。
病毒进入宿主细胞后的关键步骤之一是膜融合过程,该过程依赖病毒包膜蛋白在受体结合后的构象重排。中和抗体能够通过结合病毒表面融合蛋白的关键结构域,阻止构象转变,从而有效抑制病毒膜与宿主细胞膜的融合。
以SARS-CoV-2为例,其刺突蛋白(S)由两个亚基组成:负责受体识别的S1亚基和介导膜融合的S2亚基。S1亚基包含N端结构域(NTD)与受体结合结构域(RBD);当RBD与宿主受体ACE2结合后,S蛋白在S1/S2与S2′位点依次被裂解,触发S2亚基发生显著的构象变化,进而暴露融合肽并启动膜融合。利用这一机制的中和抗体可通过两种方式发挥抑制作用:其一,通过结合RBD阻断其与ACE2的相互作用,从而防止构象变化的发生;其二,通过直接结合S2关键融合结构域,稳定病毒处于融合前的亚稳态构象,使其无法完成膜融合所需的结构重排。
长期以来,抗体被认为主要在细胞外环境中发挥免疫保护作用。然而,研究表明抗体也能在细胞内有效中和病毒,阻止感染的进一步发展。胞质内中和机制通常发生在病毒已被内吞或部分进入宿主细胞后。抗体可与进入细胞的病毒复合物共同被内吞,在酸性内体中阻断病毒脱壳或核酸释放,从而中止感染。类似的机制多被报道于西尼罗河病毒(West Nile virus)与登革热病毒(Dengue virus),中和抗体先在细胞外结合病毒表面蛋白,病毒连同抗体复合物被内吞进入酸性内体后,抗体继续“锁定”包膜蛋白,使其无法在酸性环境中完成融合构象转换,从而导致病毒被困于内体并最终被溶酶体降解。
此外,抗体还可在细胞质内通过与胞内受体相互作用发挥效应。例如,抗体与细胞内受体TRIM21结合后,可触发蛋白酶体途径降解病毒成分,从而实现对病毒的直接清除。这类机制表明,抗体的中和作用并不局限于细胞外,而是能够延伸至病毒生命周期的多个阶段,包括进入后阻断、胞质清除等过程,构成了更全面的体内抗病毒防御体系。
抗体依赖性细胞毒作用是中和抗体在体内发挥保护效应的重要机制之一,属于典型的Fc介导免疫反应。该机制主要通过抗体将自然杀伤细胞(NK细胞)等细胞毒性效应细胞招募至被感染靶细胞。
在病毒感染过程中,中和抗体识别并结合受感染细胞表面表达的病毒抗原,其Fc片段被效应细胞表面表达的FcγRIIIa(CD16a)受体识别,触发信号级联反应。NK细胞随之释放穿孔素与颗粒酶,穿孔素在靶细胞膜上形成孔洞,颗粒酶进入细胞诱导程序性凋亡。
通过这一机制,抗体可清除受感染细胞,阻断病毒复制与释放。ADCC的效应强度与抗体亚类(主要为IgG1与IgG3)、Fc糖基化状态以及Fc–FcR亲和力密切相关。
抗体依赖性细胞吞噬作用是由吞噬细胞(主要为巨噬细胞与中性粒细胞)介导的清除机制。在病毒感染过程中,抗体结合病毒颗粒或受感染细胞表面抗原后,其Fc片段与吞噬细胞表面Fc受体(FcγRI、FcγRIIA、FcγRIII)相互作用,激活吞噬信号。被激活的吞噬细胞形成伪足包裹目标,形成吞噬体,随后与溶酶体融合形成吞噬溶酶体。在该结构内,病原体被溶酶体酶和活性氧降解销毁。
ADCP不仅有助于清除游离病毒,还能去除表达病毒抗原的受感染细胞,从而在体内维持持续的病毒控制。其效应受抗体浓度、亚类、Fc受体表达谱及糖基化状态影响。
补体活化是由一系列血浆蛋白组成的免疫级联反应,可通过中和抗体启动并介导病毒的裂解与清除。在经典通路中,抗体结合病毒表面抗原后,其Fc区域与补体蛋白C1q结合,触发补体级联反应,最终形成膜攻击复合物(MAC),直接裂解病毒包膜或感染细胞膜。此外,抗体也可通过替代通路和凝集素通路间接参与补体激活。补体系统不仅能通过直接溶解病毒或感染细胞发挥作用,还可通过“调理作用”增强吞噬细胞识别抗体-病毒复合物的能力。抗体介导的补体活化受多种因素影响,包括抗体同种型(IgM是最强的补体激活因子,IgG1与IgG3次之,IgA可通过凝集素通路激活)、抗体亲和力、价数、Fc糖基化状态以及病毒表面抗原密度和分布等。
中和抗体是宿主抵御病毒感染的核心组成部分,其功能远不止于阻断病毒入侵。近年来的研究表明,中和抗体能够在病毒生命周期的多个阶段发挥作用——从结合病毒刺突蛋白阻断受体结合与膜融合,到在内体阶段“锁定”包膜构象、在胞质内通过TRIM21途径介导病毒降解;与此同时,Fc依赖的免疫效应如ADCC、ADCP与补体激活等机制则进一步扩展了抗体的抗病毒潜能。这些多层级、多机制的防御网络,使中和抗体不仅是评估疫苗免疫原性的重要指标,也是抗病毒治疗的重要靶点。
未来的研究将更多聚焦于解析中和抗体的结构基础与免疫功能的协同关系,阐明不同病毒间的交叉中和机制,并探索如何通过工程化手段优化抗体的亲和力、广谱性与体内效应。随着单克隆抗体治疗、抗体药物工程以及疫苗设计技术的持续发展,中和抗体研究必将继续在传染病防控与免疫治疗领域发挥关键作用。
20世纪人们将电镜技术与免疫学方法相结合,逐步形成了免疫电镜检查术。随着胶体金标记技术的发展,该技术逐渐被引入到免疫电镜技术中来。胶体金是用于免疫电镜的最佳标记物,因为它呈球形,非常致密,在电镜下具有强烈反差,容易追踪在电镜下检出抗原抗体复合物。胶体金免疫电镜技术已成为目前最常用的免疫细胞化学方法之一,它具有灵敏度高、特异性强、定位精确等优点,同时它还可以对抗原进行定性、定量、定位的分析与观察。应用免疫金双重或多重标记法,可将形态、功能和结构的研究融为一体,有助于了解同一组织或细胞内不同分子间的相互关系,以及它们的合成、分泌、转运等代谢过程。如通过直接电镜、免疫电镜、胶体金免疫电镜技术等3种不同电镜技术观察新城疫病毒,结果证明用胶体金免疫电镜技术观察时由于视野中病毒粒子周围有胶体金颗粒附着,金颗粒呈现强烈的反差,很容易找到病毒粒子,免疫金标记可以显著的提高样品的病毒检出率。再如建立用免疫胶体金电镜检测技术检测犬细小病毒,证明免疫胶体金电镜技术有助于提高CPV检出率和特异性。
近年来金标记被引入生物传感器中的应用研究增多。已有许多致力于信号放大免疫传感器的研制报道,其中应用较多的是通过阻抗信号的变化来检测抗原(抗体)的含量。研究人员通过交流电导法检测了在叉指电极上的金标记的免疫凝集,从而实现了胶体金免疫层析试验的电化学检测方法,并且通过在胶体金上包被导电聚合物聚苯,提高电导法测定金标免疫反应的灵敏度。再如在标记胶体金标记A蛋白的基础上,制作了诊断森林脑炎的电化学免疫传感器,检测血清中的森林脑炎抗体及其浓度;基于铂金电极修饰的胶体金提高了生物传感器检测HABS表面抗体敏感性和稳定性;用基于胶体金表面等离子共振效应的电感受器测定胃蛋白酶的亲和常数。
免疫胶体金快速诊断技术,由于方便快捷、特异敏感、稳定性强、不需要特殊设备和试剂、结果判断直观,并可保存试验结果等优点,已在医学、动植物检疫、食品安全监督各领域得到了日益广泛的应用。目前,医学检验中应用的免疫胶体金快速检测技术主要有快速斑点免疫金渗滤法(DIGFA)和胶体金免疫层析法(GICA)两种方法。这两种方法都是以微孔滤膜为载体,包被已知抗原或抗体,加入待检标本后,经滤膜的毛细管作用或渗滤作用使标本中的抗原或抗体与膜上包被的抗体或抗原结合,再用胶体金标记物与之反应形成红色可见结果从而达到检测目的。
斑点免疫渗滤技术是20世纪80年代中期从固相酶免疫测定技术发展起来的较简便的固相标记免疫测定方法,该技术主要应用微孔滤膜(NC膜)作载体的免疫检测技术,先将抗原或抗体点于NC膜上,封闭后加待测样品,洗涤后用胶体金探针检测相应的抗原或抗体。通过金颗粒来放大免疫反应系统,使反应结果在固相载体NC膜上显示出来。研究表明免疫胶体金斑点渗滤法比间接过氧化物酶法更为敏感。
利用斑点免疫金染色法检测PPV抗体,对猪细小病毒病抗体最小检出量可达1.008×10-10g/mL,并且不同猪布鲁氏菌病、猪伪狂犬病、猪衣原体病、猪口蹄疫、猪瘟及猪弓形体病等的阳性血清出现交叉反应。通过用胶体金标记SPA建立检测猪瘟抗体水平的斑点免疫金渗滤法检测试纸盒与目前猪瘟的常规检测方法dot-ELISA法和间接血凝试验同时对200份猪血清进行猪瘟抗体检测比较,符合率达98.14%和98.19%,说明该法特异、灵敏。特异性地检测猪流行性乙型脑炎病毒抗体的斑点金免疫渗滤测定法,与血凝抑制试验(HI)对比检测了54份猪血清临床样本,两种方法的阳性符合率为71.14%、阴性符合率为65.15%、总符合率为81.14%。应用胶体金标记纯化的兔抗沙门氏菌抗体-1,SPA包被硝酸纤维素膜作对照点,建立直接检测沙门菌的斑点免疫金渗滤法(DIGFA)检测试纸盒最低检测量为4.5×106CFU/mL,将该法与国标法检测效果比较,符合率达100%。对布鲁氏菌病监控地区重点人群和不同职业人群采用斑点金免疫渗滤法(DIGFA)和试管凝集试验(STA)检测结果表明重点人群两种方法检测抗体阳性符合率为96.66%,阴性符合率为99.92%;不同职业人群两种方法检测阳性符合率为96.36%,阴性符合率为99.92%。该法敏感性和特异性与ELISA法具有良好的相符性,且具有反应快、操作简便、结果易于观察等优点,标记好的诊断试剂盒可以长期保存,随时使用,更适合于基层推广应用。
金免疫层析技术是20世纪90年代初在免疫渗滤技术的基础上建立的一种简易快速的检测技术,由Beggs等最先用于人绒毛膜促性腺激素(HCG)的测定。近年来该技术发展迅速,在生物医学领域特别是医学检验中得到了广泛应用。
胶体金快速诊断试纸条主要是将特异性的抗原或抗体以条带状包被在NC膜的检测线T上,胶体金标记的抗原或抗体吸附在结合垫上,当待测样品加到试纸条一端的加样孔上后,通过毛细作用向前移动,溶解结合垫上的胶体金标记的抗原或抗体,再移动至包被的抗原或抗体的检测线处,如果样品中含有相应的抗体或抗原,包被在检测线上的抗原或抗体和胶体金标记物与样品中的相应抗体或抗原结合,形成免疫复合物,胶体金富集在检测线处形成一条可见的紫红色线。如果待检血清中没有相应抗体,胶体金标记物将不会与包被在检测线上的抗原或抗体结合,胶体金不会富集,检测线上不会出现紫红色色线。当样品与溶化了的胶体金标记物继续往上移动至对照线时,就与包被在对照线处的特异性抗体或抗原结合,在对照线上形成免疫复合物,出现一条胶体金富集的紫红色色线。
在实际应用上,有研究人员利用猪瘟金标免疫层析试纸测试30份标准阳性血清和18份标准阴性血清,结果阳性、阴性符合率100%。与ELISA试剂、dot-ELISA试剂及正向间接血凝进行比校试验,检测血清样品205份,结果阳性率ELISA为86.8%,金标层析法为85.4%,dot-ELISA为81.5%,正向间接血凝为58.5%。应用检测口蹄疫(O型)抗体水平的免疫胶体金快速检测试纸法,与口蹄疫正向间接血凝抗原法对300份猪、鸡、鸭、牛、羊等血液或血清进行口蹄疫抗体水平检测试验,符合率达100%。这种检测技术具有操作简单快速,特异性、敏感性好,可单份测定,结果直观,且可保存试验结果,无须特殊仪器等优点,适合于广大基层单位、医院、野外作业人员以及大批量时间紧的检测和大面积普查等,因此该技术具有巨大的发展潜力和应用前景。
抗体和T细胞受体(TCR)共同构成了脊椎动物获得性免疫的两大核心。目前,单克隆抗体已成为癌症治疗、自身免疫性疾病、炎症性疾病、感染和神经系统疾病等多种疾病的主力药物之一,而针对治疗性TCR的发现、工程化改造方法,以及对其功能和药理学上的理解,发展仍比单抗领域滞后约二十年。
尽管如此,我们有充分的理由期待,基于TCR的药物未来将在多种疾病(尤其是癌症)治疗中扮演同样重要的角色。与抗体药物类似,基于TCR的药物在发现、特异性、药代动力学及最佳给药策略等方面仍面临关键挑战,必须克服这些障碍才能充分释放其治疗潜力。
与此同时,嵌合抗原受体(CAR)T细胞疗法近几年在治疗血液系统恶性肿瘤方面也取得了显著成功,极大地激发了人们将其应用于实体瘤治疗的兴趣。CAR分子通常靶向细胞表面蛋白,其设计多基于抗体结构。而基于TCR的药物则靶向细胞内抗原经处理并提呈在细胞表面的肽-MHC复合物,这一特性使其对肿瘤表面特异性抗原稀少的实体瘤具有独特的靶向优势。无论采用CAR还是TCR作为受体,一旦它们识别并结合相应的肿瘤抗原,其胞内结构域都可招募相似的信号分子,从而激活效应细胞发挥强大的杀伤作用。
目前FDA批准的所有癌症治疗单抗药物中,其靶向的细胞表面抗原均非癌细胞独有。这些抗原或多或少会在正常细胞上出现,这是癌症靶向治疗面临的根本挑战之一。而常规的αβ TCR能以极高的灵敏度和可调的特异性识别多种肽–MHC(pMHC)抗原,包括以肿瘤相关抗原(TAA)和肿瘤专属新抗原形式存在于癌细胞表面的pMHC。
最早被发现可被TCR识别的TAA包括源自MART1、gp100、MAGE-A4和酪氨酸酶(Tyrosinase)的短肽,这些抗原最初均由外周T细胞或切除的黑色素瘤病灶中的肿瘤浸润淋巴细胞(TIL)识别。与TAA类似,由癌细胞特有的体细胞突变产生的新抗原(TSA),逐渐作为TCR治疗靶向目标被认可。TIL对患者特异性新抗原具有识别能力,当这些TIL在体外被扩增后再回输,能够促使转移性实体瘤实现持久的消退,这一现象表明了针对新抗原的治疗具有巨大的潜力。此外,新抗原的多种性质(如克隆性、MHC结合特性和免疫原性)已被证明可预测免疫检查点抑制剂的疗效。由于体内产生的T细胞已具备对肿瘤抗原的特异性,这类基于TCR的疗法在癌症免疫治疗的临床开发方面引起了极大的关注。
传统抗体、类TCR抗体和TCR具有结构上的相似性,同属于免疫球蛋白超家族蛋白,但各自具有不同特征,这些特征影响了其药理学特性、潜在的应用和适用平台。
类TCR抗体和IgG在结构和药理特征上大体相同,但它们在潜在应用范围和特异性方面差异巨大:类TCR抗体能够识别包括胞内靶点在内的更广泛的抗原,但其识别的表位受到HLA限制。基于TCR的分子在识别特性上与类TCR抗体相似(尽管其亲和力通常远低于类TCR抗体),但由于天然TCR结构通常是膜结合的,其目前的平台应用受到更多限制(表1)。
传统IgG与类TCR样(基于TCR)药物之间一个重要的区别在于它们的特异性。IgG识别蛋白、糖类、半抗原等分子的三维构象,这通常能赋予其对靶抗原近乎完美的特异性。而基于TCR的药物识别的是埋藏在MHC分子沟槽中的线性肽段序列,以及邻近该肽段的MHC序列部分。因此,TCR药物所识别的表位结合表面积有限,并且产生交叉反应的可能性相当大——这既源于对MHC本身的识别,也源于其识别的肽段与蛋白质组中其他可能被呈递的肽段存在序列相似性。这种区别使得基于TCR的特异性药物的发现和开发更为复杂。通过直接从人类T细胞,如从TIL中筛选TCR,可以避免许多交叉反应,因为在T细胞发育过程中,胸腺会过滤掉大部分具有交叉反应性的TCR。
表1 免疫球蛋白超家族治疗药物的典型特征
| 特征 | IgG抗体 | 类TCR抗体 | TCR |
|---|---|---|---|
| 亚型 | 多种 | 多种 | αβ或γδ |
| 结构 | 同型二聚体 | 同型二聚体 | 异二聚体 |
| 亲和力(天然) | 高:0.1~10nM | 高:0.1~10nM | 低:0.1~10um |
| 靶抗原 | 不限 | 肽/MHC | 肽/MHC; 脂质、肽、代谢物/CD1、HLA-E、MR1; 非肽磷酸化抗原/BNT3A1 |
| HLA限制 | 无 | 有 | 有 |
| 可溶性形式(天然) | 是 | 是 | 否 |
| 膜结合(天然) | 否 | 否 | 是 |
| 半衰期(可溶性形式) | 长(数周) | 长(数周) | 短(数小时) |
| 特异性 | 高 | 多样 | 多样 |
| 目前临床适应症 | 多样 | 癌症 | 癌症 |
| 开发难度 | 简单 | 复杂 | 复杂 |
虽然TCR在结构上与单抗有相似之处,但二者在若干特性上存在差异显著,使得TCR作为可溶性药物的设计更加复杂(见表2)。因此,尽管单抗已经在多种平台上成功应用,从抗体片段到抗体偶联物再到CAR-T细胞,而TCR的可用平台迄今仍相对有限。目前已有一些单抗被发现具有与TCR相似的功能和特异性(称为类TCR抗体),这些新型单抗或许能解决TCR本身在药理学方面面临的某些难题,并大幅扩展单抗的应用范围。
表2 基于TCR疗法的障碍
| 类型 | 可能出现问题 | 可用的解决方案 |
|---|---|---|
| TCR工程细胞 | 患者自体细胞 | 采用现货型同种异体细胞 |
| TCR错配 | 使用利用CRISPR删除天然TCR的细胞;使用杵臼结构(knob-in-hole)配对设计;使用鼠源恒定区;框架区工程设计/结构域交换/单链TCR。 | |
| 免疫抑制性TME | 使用细胞因子或趋化因子装甲细胞,删除检查点分子,体外选择最佳T细胞亚群或使用细胞因子进行调节 | |
| 对肿瘤的渗透性差 | 使用细胞因子或趋化因子装甲细胞,体外选择最佳亚群或使用细胞因子进行调节 | |
| 缺乏持久性 | 使用细胞因子或趋化因子装甲细胞,过表达转录因子促进持久性或防止耗竭,体外选择最佳亚群或使用细胞因子进行调节 | |
| 供应链管理 | 自动化技术,使用现成库中细胞,包括HLA匹配的库细胞和从iPSC分化的细胞 | |
| 同种异体细胞产生的GVHD | 删除细胞中天然TCR,细胞亚群选择(如EBV/CMV致敏、CD137或CD8耗竭) | |
| 同种异体细胞产生的移植物排斥反应 | 删除HLA、B2M和其他展示机制,将HLA-E或IdeS引入细胞 | |
| 可溶性TCR | 低亲和力 | 亲和力成熟 |
| 难以进行工程改造 | / | |
| 共同障碍 | 缺乏广泛的“公共”新抗原 | 生物信息学深度挖掘,质谱驱动的实验验证 |
| 抗原异质性 | 多靶点联合,靶向必需(癌症驱动基因)靶点 | |
| 通过HLA丢失或下调逃逸 | 药物干预 | |
| 通过抗原呈递丢失逃逸 | 药物干预 | |
| 通过表位突变逃逸 | 靶向驱动突变或基本靶标 | |
| 脱靶毒性(交叉反应) | 进一步TCR计算机模拟筛查和验证性筛查 | |
| 靶向肿瘤外毒性 | 健康组织HLA配体数据库和质谱驱动的验证性筛查 |
T细胞受体(T-cell receptor, TCR)是适应性免疫系统识别并响应外来或异常内源性抗原的核心分子。αβ型T细胞所表达的TCR由α链和β链两条异二聚体组成,二者通过V(D)J重排与连接多样化产生极高的序列异质性,尤其是CDR3区域,在抗原识别中起关键作用。对个体或组织中TCR库(repertoire)的测序,能够以单分子分辨率揭示克隆组成、克隆扩增、抗原驱动选择以及免疫动力学,这些信息对基础免疫学、疫苗评估、肿瘤免疫学、移植免疫监测和自身免疫学均具有直接且不可替代的价值(例如用于追踪治疗响应、识别病理性克隆或构建表位-克隆映射)。因此,TCR-seq已成为现代免疫学研究与临床转化中不可或缺的分子工具之一。
尽管目标清晰,但不同的实验需求(如是否需要α/β配对、测序深度、样品来源、成本限制)导致了方法学上的多样化。总体上,TCR-seq可以分为两大类:bulk(群体)测序和single-cell(单细胞)测序。前者以高深度描绘群体克隆丰度分布,适合群体水平的多样性分析与与免疫反应整体趋势研究;后者在单细胞层面恢复配对的α/β信息并可与细胞转录组或表型数据整合,适合探索克隆功能异质性与细胞状态的关系。
TCR-seq早期努力依赖传统分子生物学方法与Sanger(第一代)测序。上世纪90年代至2000年代初,研究者通过克隆-分离扩增后的V(D)J片段采用Sanger测序进行少量样本的序列获取。尽管这种方式配对信息较可靠,但因通量极低、成本高昂且难以全局刻画库多样性,其应用仅限于克隆分离或小规模功能研究。此外,早期单细胞配对策略多采用手工分选单个细胞到孔板内,单细胞逆转录、嵌合PCR扩增α、β链,随后对扩增产物进行Sanger测序以得到配对链信息;该方法在能获得高度准确配对信息的同时,通量受限且工作量大。
下一代测序(Next-Generation Sequencing, NGS)的出现以及文库制备方法与分析算法的发展彻底改变了TCR-seq领域。NGS使得一次测序可以获取百万级别的CDR3序列,从而能对克隆多样性做定量分析(例如计算Shannon、Simpson指数,绘制克隆谱分布)。在bulk层面,基于multiplex PCR、5′RACE或捕获(capture)探针的文库策略广泛应用于不同样品类型(外周血、组织、FFPE等)。而在单细胞方向,高通量平台方案(如10x Genomics的Chromium V(D)J)以及基于微流控/油包水微滴或者板式分选结合Smart-seq等策略,使得在单细胞水平恢复α/β配对并与scRNA-seq联合成为可行且规模化的研究手段。近年来,长读长测序(PacBio、Oxford Nanopore)与空间转录组技术的加入,又为恢复完整可变区或在原位保持空间信息提供了新契机。总体而言,TCR测序技术的发展呈现出从低通量高准确性到高通量与多组学整合的清晰演进路径。
Bulk TCR-seq的目标是在群体水平(通常是组织或外周血样品)获取尽可能全面与定量的TCR库剖面。这类方法的核心在于如何构建代表性的文库并尽可能减少实验偏倚,使得测序得到的克隆频率能真实反映生物学丰度。
在文库构建阶段主要存在三类主流策略:多重引物扩增(multiplex PCR)、5′RACE(rapid amplification of cDNA ends)与捕获探针富集(hybrid-capture)。
多重引物法通过设计覆盖大部分V与J基因的引物集,对目标区进行PCR扩增;它的优点是对低起始量(如少量细胞或gDNA)敏感、流程成熟且成本较低,但显著缺点是引物间扩增效率不一致会引入系统性偏倚,从而影响克隆丰度的精确量化。为了缓解这一问题,常用技术包括引入UMI(unique molecular identifiers)在逆转录或接头连接时标记原始分子,以便下游去重校正PCR放大误差;或设计平衡化引物池与校准曲线来修正不同V引物的效率差异。
5′RACE则绕开对V区的特异性引物依赖,通过在cDNA 5′端添加接头实现目标扩增,能更全面地捕获V区多样性并降低引物偏倚,但对RNA质量更为敏感(RNA降解会影响5′端完整性),且实验流程相对复杂。
捕获探针法利用一组针对TCR基因组或转录本的寡核苷酸探针在文库层面对片段进行富集,这种策略在处理片段化或损坏的DNA(例如FFPE)时优势显著,且能够实现较为均一的覆盖,但试剂成本、设计与优化复杂度较高。
在测序平台方面,short-read Illumina(如MiSeq、NextSeq)仍然是bulk TCR-seq的主战场,因其高准确性、低错误率及成熟的分析生态。对于读长需求,常用2×150或2×250bp配置以覆盖CDR3区与相邻V/J序列,便于V/J注释与CDR3提取。测序深度取决于样本和研究目标:外周血样本中若目标是全面描绘多样性,常需10^5–10^6 reads/样本;若关注特定低频克隆的追踪,需更高深度或使用富集策略。
Bulk方法的优势在于成本效益与深度,可在较大样本量上做群体学比较与动力学追踪;不足之处为缺乏α/β链配对信息(通常只测β链或分别测两链),且对PCR偏倚与测序误差敏感。因此,bulk常用作群体谱系分析、克隆频率随时间变化跟踪与统计学比较,但在功能验证或需要配对信息的场景(如重构TCR进行功能测试)需要配合单细胞方法或分子配对策略。
单细胞层面的TCR测序有两类历史阶段:一代(Sanger/板式单细胞)策略与基于NGS的高通量单细胞策略。理解两者的原理与适用场景有助于选择合适的实验路径。
在NGS兴起之前,研究者常用手工或流式分选(FACS)将单个细胞分配到96/384孔板中,随后进行单细胞逆转录、利用链特异性引物或嵌合PCR分别扩增α与β链,最终通过Sanger测序获得配对序列。此方法的优点是配对信息准确、序列错误率低、每个克隆可得到完整读长,适合需要高可信度配对信息的小规模研究或功能克隆验证。缺点也显而易见,通量低、人工操作量大且费用较高,难以扩展到处理成千上万细胞的研究。
典型工作流程包括细胞单分选、采用逆转录酶进行cDNA合成、两轮或嵌套PCR扩增分别针对α/β链、琼脂糖胶电泳筛选阳性条带并送Sanger测序。该策略在早期用于从有限样本中获取功能性TCR(用于克隆构建或结构生物学研究)仍有价值,尤其当需要完整可读的V(D)J序列与高保真性时。
随着微流控与微滴(microfluidics/droplet)技术的发展,单细胞TCR-seq进入了高通量时代。现代方案可以在一次实验中处理成千至数万细胞,并在许多情况下同时获得α/β配对信息以及单细胞转录组(scRNA-seq)或表面蛋白(CITE-seq)数据。代表性平台包括基于微滴的10x Genomics Chromium,板式Smart-seq2/SMARTer改良版结合V(D)J富集,以及其他微流体或乳液方法。
以10x Genomics的V(D)J workflow为例,其基本原理是在油滴微流控系统中将单个细胞与带有条形码(cell barcode)与UMI的逆转录试剂包封到GEMs中,细胞内mRNA在油滴内逆转录并被标记上相同的cell barcode,随后对cDNA进行扩增,再通过V(D)J特异性扩增回收TCR转录本并构建文库。由于同一cell barcode下能同时捕获α与β链的cDNA,故可恢复配对信息;若在同次实验中构建了scRNA-seq文库,便能在单细胞层面将克隆身份与基因表达谱对应起来,极大地丰富了免疫细胞功能与状态的解读。
NGS单细胞方案的优势在于高通量、能直接获得配对信息并与多组学整合;但存在若干技术与生物学限制:首先,检测灵敏度有限,若某细胞的TCR转录本表达极低,可能无法被捕获;其次,微滴系统的多重包封(multiplet)会导致条形码冲突或假配对,需在下游生信中严格过滤;再次,单细胞建库成本显著高于bulk方法,且对细胞活性与处理时间要求高(影响捕获效率)。此外,单细胞方法通常捕获的是转录产物(mRNA),并不能直接反映膜表面表达或功能活性,因此常需结合功能学实验(如tetramer染色或体外刺激)进行验证。
若研究目标是对少量克隆进行高可信度的配对与后续功能学/结构学分析(例如为某一病人筛选出候选治疗TCR并进行克隆验证),一代Sanger方法仍是可靠选择。相反,当研究目标是探索克隆与细胞状态的全局关系(例如在肿瘤浸润T细胞中寻找耗竭克隆与其转录谱的联系),高通量NGS单细胞方案是更合适的路径。实际项目中,许多研究采用“先bulk筛选高丰度克隆,再在单细胞Sanger测序进行配对与功能验证”的混合策略,以兼顾广度与深度、效率与可信度。
TCR-seq技术已从单一的克隆鉴定工具演进为可同时支持群体深度剖析与单细胞层面多组学整合的成熟平台。未来的发展方向主要集中在以下几方面:首先,长读长测序(PacBio、Nanopore)有望在不借助片段拼接的情况下直接恢复完整V(D)J序列并减少注释不确定性,尤其对复杂CDR3与同源基因家族有天然优势;其次,单细胞技术将进一步与空间转录组学、蛋白质组学(如CITE-seq)与原位测序结合,以同时保留克隆信息、细胞状态及其空间分布;再次,机器学习与大规模数据库(VDJdb、McPAS-TCR等)的融合可以提高基于序列的表位预测能力,但该方向仍需大量实验验证以避免“过拟合”与错误推断;最后,标准化、参考材料与开放数据将是推动临床级别应用(如免疫监测或作为疗效生物标志物)的关键。
对于研究团队与工业用户,建议根据研究目标权衡选择:若目标是大样本量的群体学比较或长期克隆追踪,bulk(含UMI与校正)是高性价比方案;若目标需配对信息或将克隆与细胞功能直接关联,则应采用单细胞V(D)J方案并预留功能验证流程。
纳米抗体(VHH)是骆驼科动物IgG2和IgG3抗体重链的可变结构域(不含CH1结构域及轻链)表达产生的功能性片段(图1)。它们代表了最小的,能够保持原始全抗结合能力、亲和力和特异性的抗体片段(14kDa),并因其结构稳定性,可以进行抗体工程设计,制成适用于体外和体内应用的免疫试剂。相对于传统的IgG(150kDa),纳米抗体的微小尺寸赋予了与病毒隐蔽表位结合的独特能力,这一特性目前适用于生产针对病毒类的研究试剂。
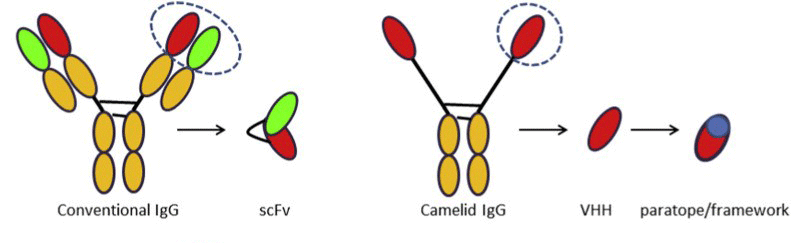
图1 传统抗体、scfv与骆驼科IgG、VHH
近年来,VHH的多功能应用场景持续拓展:其一,作为蛋白结构解析的高效晶体学伴侣及分子工具;其二,充当短半衰期放射性同位素载体,用于体内PET/SPECT成像;其三,成为纳米颗粒、纳米凝胶与生物传感器的定向免疫试剂,在特定前导肽介导下甚至可实现哺乳动物细胞内化。此外,其微小尺寸备受超分辨率显微镜结合剂研究者青睐,亦可通过串联嵌合抗原受体(CARs)实现多抗原协同结合,显著提升靶向特异性。尤为重要的是,VHH的短序列特性使其成为通过理性设计与计算机辅助优化改良生物物理特性的理想工程化抗体平台。
纳米抗体的广泛应用已推动其技术链的成熟化发展——从制备分离、表达纯化、工程改造直至最终免疫试剂开发,均建立了系统的解决方案。当前,针对不同功能需求定制化生产VHH的原核与真核表达载体体系日益完善,可精准适配各类终端应用场景。
纳米抗体通常在细菌系统中可实现功能性高产量表达,但在特定应用场景下仍需依赖哺乳动物细胞等成本更高的表达体系。例如,当需通过与Fc融合恢复体内ADCC/CDC效应功能,并利用二价分子增强表观亲和力时,必须采用哺乳动物系统以获得正确糖基化的活性分子。若Fc融合仅作为体外应用的纯化标签(如Protein A或二抗结合),则可在细菌系统中表达非糖基化VHH-Fc,其抗原结合能力依然完整保留。
对于难以在细菌中表达的融合蛋白(如辣根过氧化物酶),传统方案依赖哺乳动物细胞生产功能性纳米抗体-酶嵌合体,已成功应用于竞争性ELISA等诊断技术。当纳米抗体需在体内维持功能(如细胞内靶向治疗),哺乳动物表达系统具有不可替代性。其核心优势在于可时空精确调控表达:例如在特定生理阶段诱导荧光纳米抗体表达,实时追踪抗原动态而不干扰细胞活性;或设计竞争性抗体阻断病理蛋白聚集。此类应用依托于VHH的结构特性——其二硫键在某些构型中非必需,具有此特质的纳米抗体可在胞质内正确折叠,无需分泌途径参与天然构象形成。基于此开发的”胞内抗体”技术,通过标签-纳米抗体正交系统选择性调控靶蛋白活性,为真核细胞功能研究提供新范式。预计未来蛋白特异性纳米抗体将取代标签抗体,以这种方式更灵活地实现多维分析。
在病毒中和研究领域,哺乳动物胞质表达可精准评估表位特异性:如稳定表达抗HIV Rev蛋白的纳米抗体,通过测试其对天然Rev变体的病毒复制抑制能力,鉴定出中和关键残基并预测其对HIV-1亚型的疗效。该技术同样赋能治疗性载体开发——展示靶向肿瘤纳米抗体的重组细胞外囊泡,可实现治疗药物的定向递送。
大肠杆菌周质分泌是重组纳米抗体生产的常规策略,其氧化环境利于稳定二硫键形成。经渗透休克透化外膜后,可从上清液回收正确折叠的纳米抗体。纯化主要采用亲和层析法,针对热稳定性优异的VHH变体,还可通过热变性处理上清液实现高效回收。
该体系存在两大固有缺陷:1)分子伴侣系统缺陷易致高表达量下的错误折叠与聚集;2)有限周质空间引发的分子拥挤效应显著降低产量。
细菌胞质的还原性环境虽会抑制纳米抗体二硫键形成,但约50%的驼源VHH无需二硫键即可完成功能性折叠。此类天然无二硫键变体可通过筛选直接获得,或经分子改造(如Cys→Ser/Ala突变)实现去二硫键化。
针对含二硫键的VHH,可采用两类胞质表达策略:
分泌表达可通过培养基直接回收重组蛋白,显著降低杂质负载。该策略虽避免细胞裂解步骤,但需额外离心/过滤处理,且大体积上样延长层析时间。基于溶血素转运系统的纳米抗体分泌已实现功能性表达,但低产率制约其应用。
相较于分泌途径,包涵体作为高均一度蛋白源尚未广泛应用于纳米抗体生产。理论而言,VHH的结构特性应利于重折叠方案优化:包涵体形式表达的纳米抗体经尿素溶解与金属亲和层析后,复性效率显著提升(可达周质可溶性表达产量水平),但其构象与功能完整性仍需严格验证。最新研究表明,通过复性条件系统性筛选(如氧化还原缓冲体系梯度优化),可同步提高包涵体源纳米抗体的可溶得率及功能活性。
纳米抗体在细菌/酵母表面展示时可自发折叠为功能活性构象,该特性已广泛应用于全细胞生物传感器构建及高通量筛选。细胞表面展示技术具备三重核心优势:免纯化步骤、规避表面固定化工程、天然外向型定位。此类展示细胞可直接作为生物传感器的捕获元件或ELISA检测试剂;也可利用其溶液聚集效应实现抗原定量,相较传统”纯化-功能化”流程显著提升效率并降低成本。虽难以精确定量表面VHH密度,但可通过表达调控调整结合剂丰度。
酵母表面展示主要用于抗体片段文库淘选,通过流式细胞术对选定克隆进行分选。与噬菌体展示相比,酵母系统有巨大的优势——无需手动分选后回收克隆。基于酵母展示平台的表达革新策略,通过DTT诱导断裂展示蛋白与VHH间的二硫键,从而实现纳米抗体直接从酵母细胞中释放。纳米抗体与多种标签融合表达(如生物素化)用于SPR分析,无需亚克隆、表达和纯化。
获得纳米抗体序列后,应根据最终应用场景(如:诊断检测、结构研究、体内治疗)设计表达方案。目前广泛使用的模块化pET载体已集成以下优化设计:
纳米抗体及其融合标签的表达方式正日益多样化,以满足不同应用场景对试剂高度差异化特性的需求,这导致其生产工艺需依赖更多样的表达条件。同时,研究逐渐揭示纳米抗体的结构复杂性远超早期认知——其抗原结合位点并非单一固定构型,而是能通过框架残基参与形成多种三维构象,从而赋予抗原结合界面的多样性。尽管缺乏直接实验证据,但可推测不同VHH亚型的折叠需求可能存在差异,因此建模时必须精准选择参考结构。综上,为满足未来功能可靠性的要求,实现高度差异化的纳米抗体免疫试剂生产可能需要更多而非更少的表达系统支撑。
参考文献
de Marco A. Recombinant expression of nanobodies and nanobody-derived immunoreagents. Protein Expr Purif. 2020 Aug;172:105645. doi: 10.1016/j.pep.2020.105645.
侧向层析检测(LFA)作为纸基POC诊断领域的一种成熟策略,是最便宜、最快速且最容易使用的纸基检测,可用于现场分析/定量各种不同生物样本(包括血液、尿液、唾液和许多其他类型)中生物标志物(如蛋白质、小分子、核酸等)。LFA广泛应用于生物医学、食品污染物和有毒化学品的检测以及环境监测中。
传统商业用途的LFA存在一些局限性,包括灵敏度与实验室检测的相比较差(假阴性更多)以及特异性较低(假阳性更多)(图1A)。这些限制也阻碍了SARS-CoV-2、流感、链球菌和HIV通过LFA进行检测,维持了采用更复杂的实验室检查(如PCR、ELISA)进行诊断的要求。例如,即使有商业SARS-CoV-2 LFA,其临床检测准确性(阳性预测值:11%~50%)远低于声称的87%~97.5%的灵敏度和100%的特异性,这限制了LFA对大规模流行病的控制和管理。
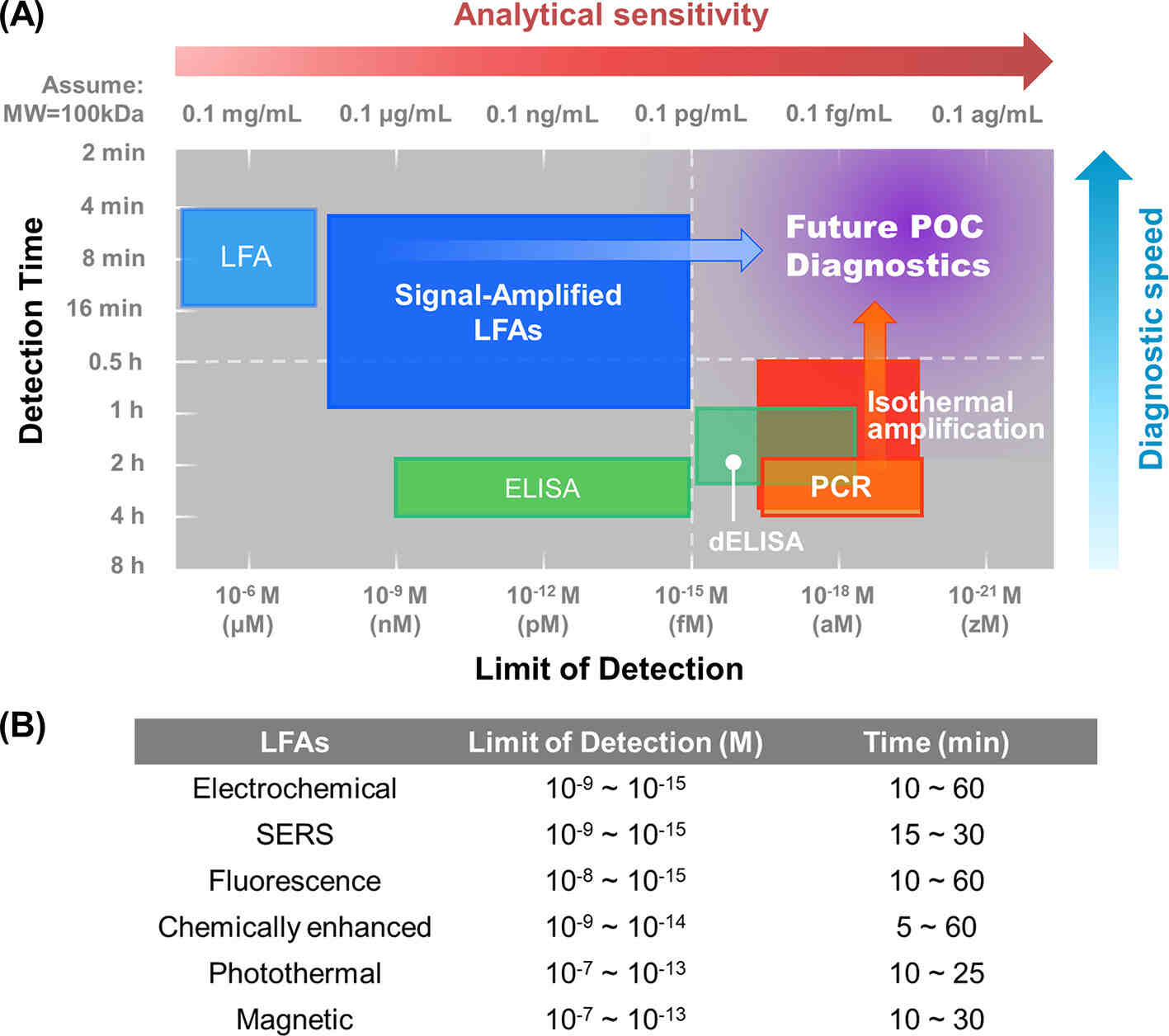
图1 信号增强LFA与其他诊断工具的分析灵敏度和诊断速度的比较
为了解决这些限制,人们投入了大量精力来提高LFA的灵敏度和特异性,以实现更准确和更高性能的POC测试。提高灵敏度的两种重要策略包括检测方法改进和样本富集。如信号增强LFA可以将检测灵敏度提高几个数量级,使LFA能达到ELISA级别的灵敏度。此外,进行样本预富集,并配合读数系统可提供PCR级灵敏度。虽然灵敏度提高了,这些技术往往需要更长的检测时间。因此,平衡灵敏度和检测时间对未来POC诊断的开发构成了重大挑战。LFA的特异性也很重要,主要通过分析优化、鉴定和使用高亲和力和高特异性试剂来提高。图2描述了夹心LFA结构以及用于提高LFA灵敏度和特异性的样本富集、分析优化和信号增强方法。
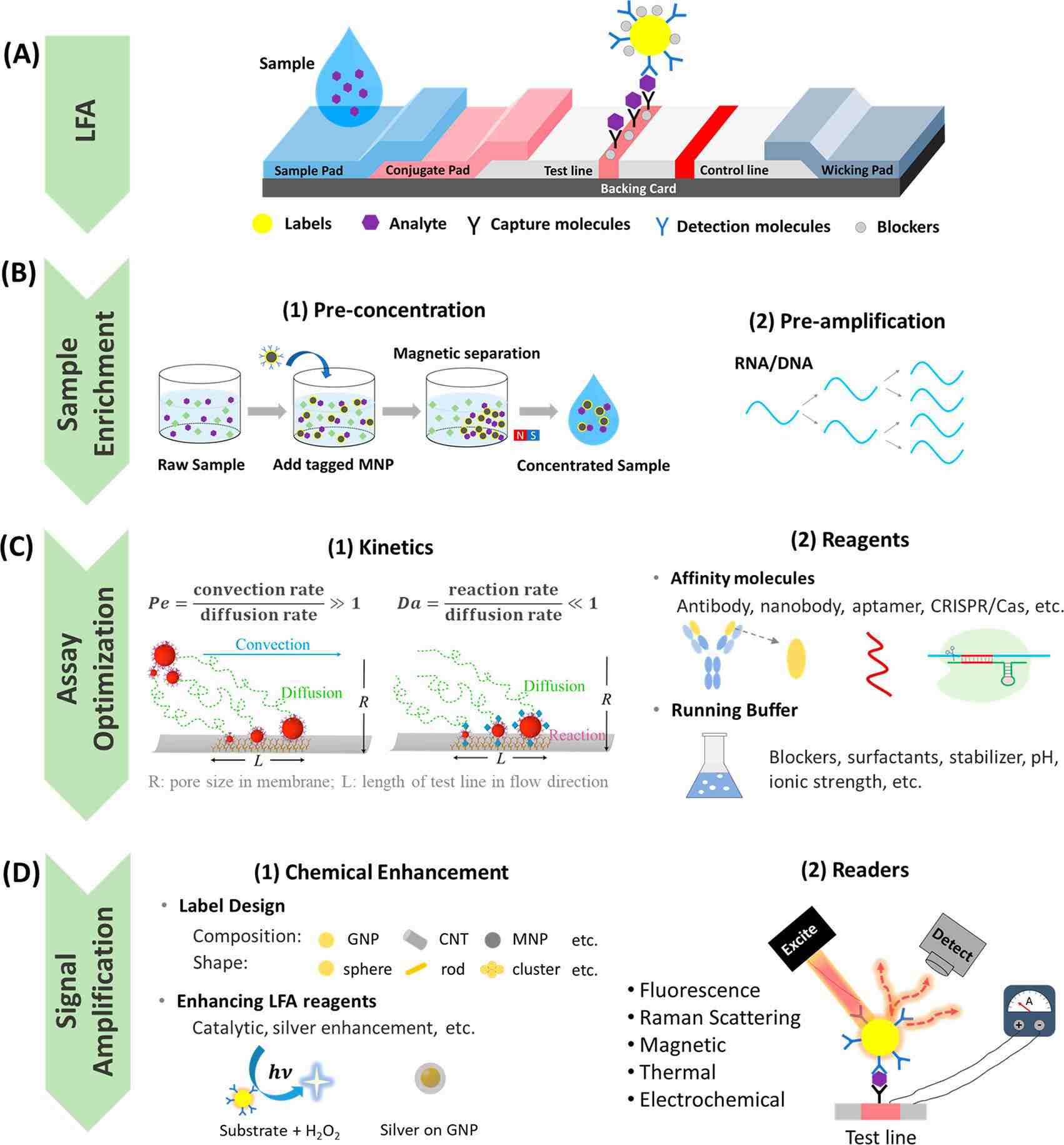
图2 提高LFA灵敏度和特异性的策略
了解检测动力学是LFA开发的基本步骤,对于提高LFA灵敏度至关重要。这最终影响特异性结合(SB)和非特异性结合(NSB),进而决定检测的灵敏度和特异性。因此,优化分析动力学的目标是最大化SB和最小化NSB,可以通过最大化信噪比(SB/NSB)来实现定量分析优化。
传输动力学和反应动力学分别用佩克莱特数(Pe)和达姆科勒数(Da)来表征,它们定义分别为:

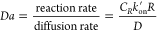
其中:
U是移动流体通过LFA膜的速度,R是膜内孔隙的特征大小,D是流体中运输的分子和结合标记的扩散率;CR是测试区域中捕获分子的浓度 ,Kon′是测试区域中偶联标记的有效正向免疫反应速率常数,假设Kon′=nKon,其中kon是单个抗体-抗原免疫反应的正向反应速率常数,n是每个标记颗粒的有效抗原数。
表1 LFA中Pe和Da的估计值
| 参数 | 估计值范围 |
|---|---|
| 膜的特征孔径 L(m) | (3-20)×10–6 |
| 平均流体流速 U(ms–1) | (0.5–3)×10–4 |
| 扩散率 D(m2s–1) | 10–12–10–10 |
| 结合速率常数kon′(M–1s–1) | 103–105 |
| 捕获抗体浓度CR(mol m–2) | 10–10–10–8 |
| 佩克莱特数Pe | 10–102(≥1diffusion limit) |
| 达姆科勒数Da | 10–4–10–1(≤1reaction limit) |
根据表1中的Pe和Da估计值,LFA受反应速率限制,提高反应效率是实现SB最大化和提高LFA灵敏度的最关键步骤。分子和标记物的传输受扩散速率的限制(≤对流速率,Pe=10–102),表面反应受反应速率的限制(≤扩散速率,Da=10–4–10–1)。反应效率的提高可以通过提高反应速率来实现,反应速率与反应速率常数(反应动力学)和反应物浓度成正比,和/或通过增加反应时间来实现。
针对夹心法检测,增加偶联物/抗原/捕获抗体三元反应动力学:当抗原首先与偶联标记物结合,然后在预混合流中与捕获抗体结合时,夹心三元形成的速度比它首先与捕获抗体结合,之后在顺序流中与偶联标记结合时慢。顺序流的检测限(LoD)值比预混合流的低4到10倍。而顺序流的缺点在于较长的检测时间。
对于核酸杂交,杂交动力学与盐浓度低于0.2 M时的离子强度密切相关。在测试区域前的膜中添加盐水屏障以加速杂交反应,在不改变LFA形式和程序的情况下,检测灵敏度可以提高10倍。这也会减慢流速,增加检测时间。
对于抗原和抗体之间的大多数免疫反应,反应速率常数相对不变。提高反应物浓度可能是提高反应速率,增加测试区域中捕获标记物数量的有效替代方法。因此,在将样本引入侧向层析测试之前,可以将样本中的分析物进行预浓缩(图2B(1))。如有研究人员在侧向层析之前,同时使用磁性分离对样本中的分析物进行预浓缩,灵敏度提高了10倍。有研究称,通过在样本中加入非离子表面活性剂TritonX-114以形成两相胶束系统,可以将预标记的分析物预浓缩到无胶束层中,该方法可将LoD提高约10倍。
其次,分析物也可以在LFA的流动周期内浓缩。例如,等速电泳有助于预浓缩抗原偶联复合物并增强LFA中的传输动力学,标记物的表面反应速率和平衡结合显著增加,LoD提高了400倍。
第三,增加偶联标记的有效结合位点的数量可以提高反应速率,从而降低LoD值。可以通过标记设计或检测分子的特定定向来实现结合位点的增加。例如,可以增加金纳米颗粒(GNP)大小,或者可以通过颗粒表面多层功能化,加载更多检测分子,从而增加结合位点的数量。值得注意的是,颗粒大小和涂层受到保持颗粒在流经多孔膜的过程中的稳定性和扩散率的限制;否则,可能会因沉淀和/或NSB而产生染色或背景噪音。与通过传统物理吸附的随机定向偶联相比,通过改进的偶联方法使检测分子在标记表面的特定定向可以产生更有效的结合。可以通过化学层介导的共价结合(如聚乙二醇化)或生物分子层介导的生物亲和结合(如protein A和protein G、生物素-链霉亲和素偶联、DNA介导的固定)来实现特异性定向。此外,应优化检测分子的覆盖范围,以最大限度地减少密集的检测分子层产生的任何位阻,并最大限度地提高对分析物的亲和力。
第四,可以增加测试区域中有效结合位点的数量。如测试线上常规捕获分子可以用3D“蛋白”探针代替,其中多个肽段是自组装定向的,增强了反应性,从而将灵敏度提高4到8倍。用纤维素纳米纤维修饰膜以加载更多捕获分子也可以将检测灵敏度提高20倍。据报道,纤维素纳米纤维使捕获分子更接近表面,从而将捕获标记的比色强度提高了36.5%。
增加反应时间可以增加测试区域中捕获的标签数量。例如将棉线放入膜中可以减慢流速,将检测灵敏度提高4倍。在膜和偶联垫之间添加堆积垫也能延长反应时间,将LoD提高1.1至2倍。
除了通过上述方法最大化SB外,最小化NSB对于提高LFA的灵敏度也很重要,因为NSB会干扰极低浓度分析物的检测,并限制灵敏度的提高。例如,40nm金纳米球(GNS)的聚乙二醇化可以减少不稳定的柠檬酸盐和稳定的NGS的聚集,帮助NSB大幅降低,从而提升检测灵敏度。同样,在金纳米颗粒上加入二氧化硅涂层,通过赋予颗粒高稳定性显著降低背景噪声。这种涂层还增加了颗粒表面积,从而能够加载更多抗体。结果表明,二氧化硅包被的GNP提高LoD30倍。综上,标记优化后的LFA灵敏度比传统的裸GNP标签高10倍。
总之,优化检测动力学是改进LFA的常用方法,LFA的灵敏度提升可多达百倍。
除了优化检测动力学外,放大测试区域的信号也可以普遍提高灵敏度。通过化学增强增加阳性测试区域的比色对比度是一种非常直接的方法,可以在保持目视检测便利性的同时增强信号。这种增强的对比度可以通过标签设计和使用增强LFA解决方案来实现。
标签设计法通常保持传统LFA形式,使用具有更强的比色对比度的标签取代传统小粒径(约20~40nm)GNS。如图2D(1)所示,可以通过优化GNP的结构和大小或使用粒子簇或由其他金属、金属氧化物或有机材料制成的颗粒替换GNP来实现更强的对比度。例如,GNP修饰的二氧化硅纳米棒标记(即包覆胶体金的微型二氧化硅纳米棒)在检测兔IgG时实现了比传统GNS LoD值低50倍。又如聚苯乙烯微珠用作纳米级GNS的载体以增强测试区域的比色对比度,将流感病毒H3亚型的检测灵敏度提高了64倍,比基于10nm GNP的LFA提高了16倍。使用碳纳米管(CNT)作为兔IgG检测的标记物,相比使用传统GNP检测限降低了3个数量级,这是由于CNT的更高纵横比,使得能够结合更多的检测抗体,提高了免疫反应速率。标记设计保留了LFA快速响应、使用简单和低成本的优势,无需改变任何检测形式或步骤。
LFA增强试剂可用于在正常测定后在测试区域诱导催化或其他化学反应,以放大比色对比度。催化反应通常是通过使用酶或纳米酶催化测试区域的氧化还原反应来实现的。使用最广泛的酶是辣根过氧化物酶(HRP)。在LFA中HRP与检测抗体耦连;上样后洗涤,去除膜上多余的标记物;HRP底物和H2O2溶液流经LFA催化,在测试区域产生强烈的显色反应或化学发光。有研究显示,通过酶促反应,检测灵敏度比传统基于GNP的LFA提高了1个数量级。
纳米酶是基于纳米材料的一类人工酶。与天然酶相比,纳米酶拥有更高的催化稳定性、更容易的修饰过程和更低的制造成本,近几年迅速发展为天然酶的直接替代物。一些纳米酶已经被用作LFA的标记物。如Pt纳米催化剂(Au@Pt core@shell结构)的使用实现了血清中p24蛋白LoD低至0.8 pg/mL,甚至优于商业ELISA的灵敏度(>1pg/mL)。
其他化学增强技术包括银增强、双金共轭和诱导金聚集。在银增强方法中,在常规测定后,通过将Ag还原剂流经LFA,Ag在测试区域中捕获的GNP上成核。GNP标签表面上生成的Ag层放大了测试区域的显色强度,灵敏度提高了约10倍。同样,双金共轭引入次级GNP与测试区域内已经捕获的初级GNP结合,从而增强显色强度。这种结合可以通过利用高生物素-链霉亲和素结合亲和力或者一抗和二抗之间的反应来实现,与间接ELISA的原理类似。诱导金聚集方法类似于双金共轭方法,但该方法可以在捕获的GNP上涂覆更多的GNP,从而更好地放大显色强度。大的聚集体运输速度较慢,因此需要的检测时间更长。值得注意的是,尽管这些方法提高了灵敏度,但由于偶联了多个反应步骤产生了不一致性,会降低量化信号(即最初捕获的标记)的能力。
除了化学增强方法外,LFA信号还可以通过使用检测仪进行放大。使用检测仪,测试区域中捕获的纳米颗粒(NP)标记被外部物理刺激(如激光、电势或磁场)激发(图2D(2)),以产生放大信号。使用光学/电/磁传感器检测相应信号可以识别背景上的微小信号差异。检测仪系统可以将检测灵敏度提高到传统的视觉读数几个数量级,可与ELISA的灵敏度媲美。此外,信号的定量(即标记量)可以通过微孔检测系统实现,因为信号强度通常与测试区域中捕获的NP数量成正比,这也与目标分析物的数量相关。根据激发方法,有多种类型的检测仪可供选择,包括荧光、表面增强拉曼技术(SERS)/光热法(即热对比度、光声成像/光热激光散斑成像/和热光子锁相成像)、电化学和磁放大。
以上方案在灵敏度优化上取得了实质性的进展,但仍需要综合分析,以减少同一技术内的性能差异。优化后的LFA灵敏度比传统商业LFA高1~9个数量级,基本可以达到ELISA的灵敏度(图1A)。部分技术的灵敏度,如荧光LFA和纸质电化学诊断,甚至可以达到fM水平(ELISA灵敏度的下限)。同时,我们需要注意的是,同一技术的LoD存在很大差异(3~6个数量级),这种差异是由于分析优化不足造成的。在检测过程中,NSB不可避免地与SB共存。当进行信号放大时,信号(来自SB)和噪声(来自NSB)都被放大;因此,假阴的数量减少了,但假阳的数量可能增加。换句话说,灵敏度的提高是以牺牲特异性为代价的。特别是对于低靶标浓度,NSB捕获的标记量可能与SB捕获的标记量相当,甚至更大。在这种情况下,进一步增加外部激励场的强度无法区分信号和噪声,而会产生假阳。因此,信号放大的效果可能会受到标记物NSB的限制。
为了解决这一限制,需要一种与信号放大技术兼容的综合分析优化方法(见图3)以降低NSB。经过这种优化后,可以使用更强的外加场或增强LFA试剂来进一步提高检测灵敏度,直到特异性受到影响(即假阳性的数量增加)。这种信号放大和分析优化的方法可以多次迭加进行,直到获得满意的灵敏度和特异性。
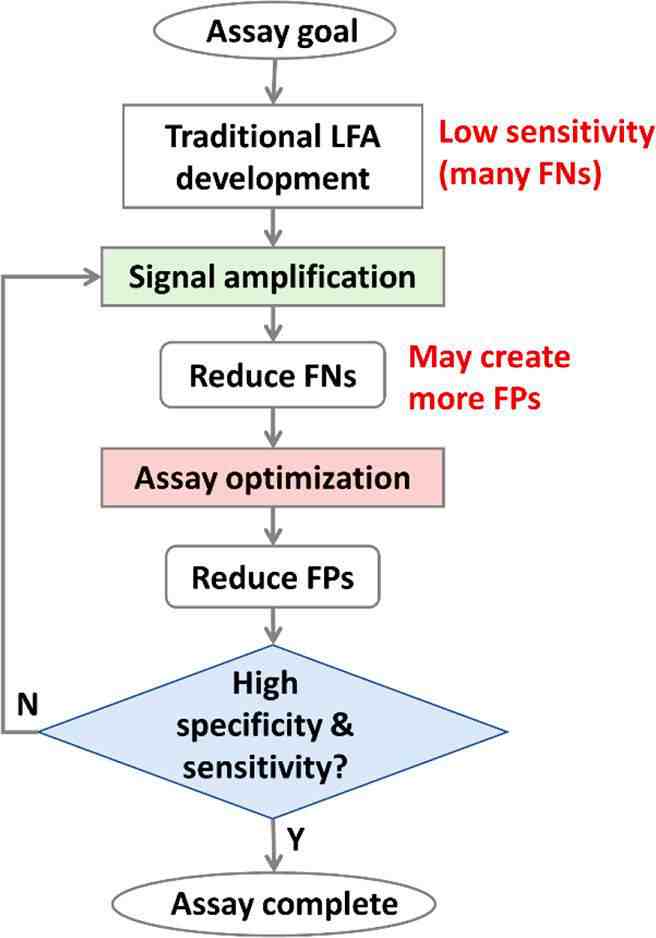
图3 结合信号放大方法进行综合分析优化
除了灵敏度之外,检测速度、易用性和成本也是评估POC诊断应用技术的重要指标。表2总结了LFA的优缺点和未来必要的改进,图1B显示了它们的检测时间的详细比较。如表2所示,化学增强保持了快速视觉检测的优势。相比之下,使用增强型LFA试剂会显著增加检测时间、检测复杂性和不精确性,因为需要额外的步骤来递送各种试剂。检测时间跨度从20分钟至1小时不等。为了解决检测时间增加的问题,研究人员试图通过设计一些LFA装置来实现多溶液自动化递送。同时,可以通过实现装置小型化,自动化所有检测步骤以及通过探索反应动力学来缩短检测时间。
表2 LFA的优点、缺点和未来改进的方向
| 信号放大 | 优点 | 缺点 | 未来改进的方向 |
|---|---|---|---|
| 化学增强 | 提高灵敏度 | 提高NSB | 自动化/小型化/操作简易化 |
| 快速读数 | 大标签的扩散问题 | 综合分析优化 | |
| 增加时间和复杂性 | 检测多种分析物 | ||
| 可能无法量化 | 多方法连用 | ||
| 创新的信号处理方法 | |||
| 检测仪 | 提高灵敏度 | 提高NSB | 临床验证 |
| 指标量化 | 增加了步骤和时间 | 小型化 | |
| 高重复性 | 增加成本 | 成本控制 | |
| 结构复杂(电化学) | 综合分析优化 | ||
| 检测多种分析物 | |||
| 多方法连用 | |||
| 创新的信号处理方法 |
同样,使用检测仪也会增加检测时间和分析成本。大多数检测仪的检测时间通常在20分钟以内,电化学法通常在30分钟以内,但是一些多步骤的测定可能需要1小时以上。研究人员做了诸多努力来减少检测仪的扫描时间。例如,最快的SERS检测仪可以在5秒内扫描一个测试区域,并且正在开发扫描时间<1分钟的热敏电阻检测仪。检测仪性能的可靠性和可重复性也仍然值得探究。未来需要进一步开展小型化、降低成本和临床验证工作,以使这些检测仪更适合POC应用。
此外,由于可以降低成本、样本量需求较小以及能够快速区分具有相似症状的常见疾病等优势,研究人员开始转向开发能够同时检测多种分析物的多重LFA。多重LFA存在一些挑战:包括交叉反应性、LFA试纸多重测试区域的物理限制以及临床验证存在困难。
最后,可以使LFA实现亚飞摩尔级检测灵敏度的新的信号处理方法已经出现。LFA信号通常以模拟信号形式读取,这本质上限制了灵敏度提高的上限,即检测仪需要累积数百万个标签才能产生可检测的信号强度,这很难实现类似于ELISA亚飞摩尔级灵敏度。相比之下,数字ELISA(dELISA)采用数字信号的采集方法,可以计数免疫反应后每个酶标信号,检测到亚飞摩尔级至渺摩尔级浓度。然而这些平台尚未准备好用于POC,其制造和使用的复杂性或信号处理所需的周转时间很长。尽管如此,考虑将dELISA的信号采集方法与现有的LFA相结合,开发一种更便宜、更简单的数字检测平台用于超灵敏POC诊断是有价值的。综上,经过分析优化、设备升级和验证,以上方法有望适应未来的超灵敏POC应用。
参考文献
Liu Yilin, Zhan Li, Qin Zhenpeng, et al. Ultrasensitive and Highly Specific Lateral Flow Assays for Point-of-Care Diagnosis[J].ACS Nano 2021 15 (3), 3593-3611.DOI: 10.1021/acsnano.0c10035.
近年来,抗体药物凭借其高靶向特异性、低免疫原性和长效药代动力学特性,在多种疾病治疗领域获得广泛应用,获批上市数量显著增加。得益于现代生物技术的进步,抗体发现平台日益成熟,具备特定功能的生物大分子持续涌现。然而,抗体药物研发仍面临较高的失败率,导致最终获批药物有限。其核心挑战在于分子固有的易聚集性、稳定性不足和溶解性差等特性,这些缺陷为后续的生产工艺、制剂开发、储运环节带来显著困难,阻碍了药物成功开发。因此,在早期分子筛选中,亟需运用体外方法评估分子的均一性、稳定性、溶解度和结合特异性,筛选出优势候选分子,并识别其潜在风险。这一过程——成药性评价——旨在降低因分子自身不良属性导致的后期研发失败风险。
成药性评价主要通过一系列高通量的技术手段,对抗体的一级结构、翻译后修饰、聚体和片段、电荷异质性、疏水性、构象稳定性、胶体稳定性、溶解度、黏度、蛋白质-蛋白质相互作用、非特异性相互作用、表位竞争和亲和力等方面进行多维度的分析,筛选出最佳的候选分子,确保在后续的开发中具有较为稳定的性质,从源头上保证产品的质量。
传统上,抗体药物发现阶段的筛选过程被认为是漏斗型筛选,通过杂交瘤技术、展示技术(噬菌体展示、细胞表面展示、无细胞分子展示技术)、人源抗体转基因小鼠和人杂交瘤技术、展示技术(单B细胞测序等技术,获得数量庞大的候选序列,以与目标种属蛋白特异性结合、功能活性等评价方法作为主要评价策略进行多轮筛选。但目前越来越多的研究者将成药性评价提前至抗体药物发现阶段,有利于尽早评估和解决候选药物的可开发性问题,最终获得一个均一、稳定、安全、有效和可放大生产的候选序列,提高药物开发的成功率和效率。
抗体药物早期发现阶段的成药性筛选流程划分为成药性初期评价、成药性筛选和成药性评估3个阶段。成药性初期评价和筛选阶段,样品种类多、数量大,综合时间进度和有限的资源,采用倒漏斗型筛选策略,选择少量且关键的质量属性进行评价,同时配合高通量技术加快评估流程;对中后期筛选出的分子,应进行多维度且较为全面的质量属性的表征;对最终筛选出的分子,应增加加压实验等研究,以获得更为全面的分子属性。值得注意的是,在抗体药物发现阶段,成药性筛选评估没有绝对的标准对候选分子进行剔除,而是对候选分子综合评估后,打分排序(如风险等级按高、中、低排序),帮助及时了解候选药物的分子属性和潜在风险因素,掌握控制要点,便于后续工艺和制剂的开发。
非人源的抗体候选序列(如杂交瘤筛选技术)获得后,需先构建为嵌合抗体,即对候选序列进行恒定区人源IgG1或IgG4代替,使用哺乳动物细胞系(人源胚胎肾细胞、中国仓鼠卵巢细胞等)表达;对于筛选获得的人源序列,可以直接进行质粒构建和表达;对于抗原结合片段库(Fab)、单链抗体可变区基因片段库(scFv)和单域抗体库(dAb)筛选得到的序列,可以制备成IgG-like的形式,也可以选择合适的表达体系进行单独表达。此阶段的表达方式一般为瞬转表达,有利于快速获得小体系样本,经高通量设备纯化,得到具有一定纯度的样品。
此阶段是以活性筛选策略为主,包括结合活性[酶联免疫吸附技术(ELISA)、流式细胞技术(FCM)]和功能活性(天然细胞、工程细胞),还可以借助高通量分子相互作用仪,如基于表面等离子共振技术(SPR)的BiacoreTM 8K系列仪器或者基于生物膜干涉技术(BLI)的Octet® BLI系列仪器,检测与目标种属靶抗原的特异性、亲和力、结合动力学和表位竞争等。在保证项目进度的前提下,可选择关键物理化学属性进行成药性初期评价,包括表达量和纯度。纯度可通过高通量技术进行检测,如体积排阻色谱-高效液相色谱(SEC-HPLC)法、十二烷基硫酸钠-聚丙烯酰胺凝胶电泳(SDS-PAGE)法或毛细管凝胶电泳-十二烷基硫酸钠(CE-SDS)法等。瞬转表达量与稳转表达量具有一定的相关性,筛选高表达量的分子,同时选择不易形成聚集或降解的分子,不仅可以降低后期生产成本,还代表着具有更优的构象稳定性和胶体稳定性,也为体外活性检测奠定基础,避免因聚集体或降解物过多导致结果失真。
筛选出的序列若用于制备双特异性抗体,应在此阶段进行组装考察,评价活性和稳定性,可以通过构建质粒表达,也可以借助体外重组技术完成。评估项目包括表达量、纯度和活性,值得注意的是:纯度检测项应根据分子及其变异体性质的差异选择合适的评价策略,如可根据电荷差异通过离子色谱-高效液相色谱(IEX-HPLC)和成像毛细管等电聚焦(iCIEF)等方法进行表征,也可根据分子量大小通过SEC-HPLC和毛细管凝胶电泳法表征。
筛选出的序列若用于制备抗体偶联药物,可在此阶段进行偶联,评价分子偶联效率和稳定性,如通过SEC-HPLC等方法检测聚体和片段的含量、通过疏水相互作用-高效液相色谱(HIC-HPLC)法或质谱法检测ADC的偶联量及偶联位点的分布、通过质谱法检测小分子脱落量和通过ELISA法或质谱法检测血浆稳定性等。
经以上步骤筛选出的分子,借助高通量计算机模拟和分析技术,对氨基酸序列、分子结构、蛋白质和蛋白质相互作用进行初步分析和预测。针对抗体中的非人源性成分,进行人源化改造,以降低在人体内出现免疫原性的风险,保留抗体互补决定区非人源氨基酸残基,其余均替换为人抗体的相应部分,或改造暴露在抗体表面的骨架区的非人源氨基酸残基;对于全人源抗体库筛选出的人源抗体,需进行亲和力成熟等改造。不同的突变改造可能会影响分子稳定性,应综合考虑平衡亲和力、免疫原性和分子稳定性;对于其他潜在的一些问题,如等电点、脱酰胺、异构化、暴露的色氨酸和蛋氨酸、未配对半胱氨酸、糖基化或糖化等也应给予关注和改造,如等电点过高可能会影响半衰期,等电点过低在常规的工艺过程中易产生聚集;互补决定区的脱酰胺、异构化导致分子降解或者活性降低;氧化可能会影响活性,也可能引发聚集体的产生;可变区的糖基化或糖化位点,可能会影响活性和稳定性等等。
改造后的分子通过轻重链的组合配对,产生一系列的变体,数量一般在100个左右,通过哺乳动物细胞瞬转表达和高通量纯化(依据活性实验需求进行内毒素控制),评价表达量、纯度和生物学活性,去除极差分子后,开展成药性筛选评价。
成药性筛选阶段的目标为对各分子的固有属性进行评估和排序,如电荷变异体含量、疏水性、热稳定性、不同粒径聚集体、自相互作用、非特异性相互作用、与不同种属抗原的亲和力、表位差异、Fc功能等。最终挑选出具有较低异质性、较高稳定性、较长半衰期、高产量和可工艺放大的抗体,筛选出的分子数量一般小于5个。本阶段成药性筛选所用到的检测技术,应具备样品用量少、通量高、分析快速等特点。若候选分子具有特殊属性,可在此阶段增加初步的稳定性评估,以确保样品在检测、存储、动物给药过程中的稳定,如室温稳定性、2−8℃稳定性、冻融稳定性等。若候选分子计划用于制备高浓度剂型,可以增加溶解度预测实验,随着蛋白浓度的升高,相较于低浓度溶液,蛋白与蛋白之间的作用力增强,但传统的溶解度测定方法对蛋白消耗量大,故可选用在较低浓度中预测溶解度的方法。
对于最终筛选出的几个候选分子,进行进一步的可开发性评估。表达阶段扩大瞬转体系,纯化阶段增加精纯步骤,并严格控制内毒素含量,为早期的动物体内药代动力学(pharmacokinetics, PK)、药效动力学(pharmacodynamics, PD)以及毒理研究储备样品。成药性筛选阶段进行的成药性表征方法应再次检测和确认,另外还应进行加压实验的考察,有助于快速地暴露分子的风险位点及变化趋势,考察对抗体特异性和功能活性的影响程度,考察分子的耐受性和可及性,评估敏感条件,预测长期稳定性,如设置40℃高温稳定性,纳米抗体可进行100℃高温稳定性考察。通过翻译后修饰、聚集体和降解物含量、酸碱变异体含量、ADC中小分子脱落量的变化,发现分子潜在的翻译后修饰位点、降解位点及变化趋势;还可以设置强酸、强碱、氧化和光照等加压实验;对于高浓度制剂的开发,在前期溶解度预测实验中,由于高的稀释因子,难以准确推导高浓度制剂真实的溶解度和黏度,故在此阶段,可以开展目标浓度的浓缩和过浓缩,使用常用缓冲体系,添加尽可能少的制剂辅料,观察药物在浓缩过程中的特性,包括但不限于蛋白含量、聚集体和降解物的含量、粒径的尺寸分布和分散度、黏度、热稳定性、储存稳定性以及在高浓度下与血清的相融性等。最佳配方条件的探索,则应在后期制剂开发中进行。
成药性评价作为一种在药物发现阶段被广泛认可的候选分子筛选方式,已经成为抗体药物研发过程中的关键一环。不同成药性筛选阶段,选择符合筛选目的的评估策略,不仅能理解分子目标属性,还能大幅减少筛选时间,降低冗余数据堆砌,最终加快筛选流程和提高研发成功率。
参考文献
姚江宁, 律璎桐, 张迎珺, 张正平, 徐同杰. 抗体成药性筛选流程及评价策略[J]. 生物工程学报, 2024, 40(2): 507-516
T细胞受体(T cell receptor, TCR)是适应性免疫系统中识别抗原的核心分子。与抗体类似,TCR能够通过高度特异性的方式识别抗原,但不同之处在于TCR只能在主要组织相容性复合体(major histocompatibility complex, MHC)分子呈递的背景下识别展示在机体自身细胞表面的抗原。正是这种“肽–MHC”复合物(pMHC)的特异性识别,使得TCR在清除病毒感染、肿瘤免疫监视以及免疫耐受的建立过程中扮演不可替代的角色。
本文将介绍TCR是如何通过独特的结构基础,实现对抗原的高特异性识别的。通过对分子结构的梳理,我们可以更好地理解TCR与抗原之间的相互作用机制,以及这些结构特征如何被应用于免疫治疗、疫苗设计和疾病诊断。
TCR是由通过二硫键连接的两条多肽链组成的细胞表面异二聚体。根据链的组合类型,TCR可分为两类:αβ-TCR,是大多数T细胞识别抗原的受体,主要识别由经典MHC I类分子(如HLA-A/B/C)或MHC II类分子(如HLA-DR/DP/DQ)提呈的抗原肽;γδ-TCR,可直接识别非肽类抗原(如磷酸化代谢中间产物、脂质)或应激分子,无需MHC分子提呈,具有更广谱的抗原识别能力。
TCR在结构域组织和结合模式上与抗体Fab片段具有相似性,每条TCR链均由可变区(variable region, V)和恒定区(constant region, C)的Ig样结构域、跨膜区及短胞质尾区组成。
在可变区中,最关键的部分是互补决定区(complementarity-determining region, CDR)。αβ-TCR通过其CDR与pMHC复合物结合,其中CDR1和CDR2主要来源于种系V基因片段,通常与MHC分子接触;而CDR3则由V(D)J重排过程中产生的多样性序列构成,是与抗原肽直接相互作用的主要区域。
在跨膜区及胞内区域,TCR自身的胞内尾巴极短,无法直接进行信号转导,因此与CD3复合物共同组装,形成完整的信号复合体。跨膜域通过带电氨基酸与CD3形成稳定相互作用,而信号转导则依赖于CD3分子内含的免疫受体酪氨酸活化基序(ITAM)。当TCR识别到抗原肽–MHC复合物后,这些ITAM会被Src家族激酶磷酸化,从而启动下游的T细胞激活级联反应。
总体而言,TCR的结构设计可以概括为“三段式”分工:可变区负责识别,恒定区提供稳定支架,跨膜区和胞内复合物负责信号转导。正是这种结构上的层层配合,使得TCR在保证特异性识别的同时,还能迅速转化为细胞内的信号,从而触发免疫效应。
TCR的核心功能在于其对抗原肽–主要组织相容性复合物(pMHC)的特异性识别。MHC I类分子通常呈递来源于细胞内蛋白降解的肽段,主要被CD8⁺细胞毒性T细胞识别;MHC II类分子则主要呈递细胞外摄取的肽段,由CD4⁺辅助性T细胞识别。这种分工使得免疫系统能够覆盖病毒感染、肿瘤突变和外源病原体等不同来源的威胁。
在TCR–pMHC的对接模式(docking mode)上,研究表明大多数αβ-TCR与pMHC之间呈现相对固定的倾斜角度,称之为对角线结合模式(Diagonal docking mode)。其特点是TCR以一定角度覆盖pMHC结合槽的长轴上。在该模式下,TCR的CDR1与CDR2主要与MHC分子的α螺旋结构形成接触,而CDR3则深入抗原肽的核心区域,决定结合的特异性和多样性。这种结合模式保证了TCR对MHC框架的限制性识别,同时通过可变的CDR3区域与抗原肽直接接触,形成高度特异性的识别界面。虽然存在一定的例外情况,但这种“对角线”结合模式在结构研究中被反复观察到。
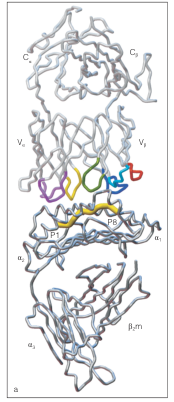
图1 TCR β链的CDR1、CDR2和CDR3环分别为浅蓝色、深蓝色和绿色;α链的CDR1、CDR2和CDR3环分别为浅紫色、深紫色和黄色。β链HV4环为红色。黄粗线P1-P8是结合肽。
结构证据来自经典的X射线晶体学研究。例如,Garcia等人在1996年解析了第一个人类TCR–pMHC复合物的晶体结构,揭示了CDR1、CDR2环与MHC分子α螺旋的相互作用模式,以及CDR3环深入结合抗原肽的关键作用。随后Rudolph等人的工作进一步总结了不同TCR–pMHC复合物的结构共性与差异,强调了TCR在兼顾MHC限制性与抗原多样性方面的分子基础。这些发现奠定了我们今天对TCR抗原识别机制的理解。
虽然TCR可与pMHC复合物形成高度特异性的结合,但其胞内区极短,无法直接介导信号转导。TCR的信号功能完全依赖于CD3复合物的参与。CD3复合物由CD3γε、CD3δε以及CD3ζζ二聚体组成,含有多个免疫受体酪氨酸活化基序(ITAM),是信号放大的关键元件。
在跨膜区的分子互作上,TCR的α链和β链跨膜螺旋区域带有带电氨基酸残基,分别与CD3跨膜结构中的相反电荷残基形成静电互补。这种“电荷对”互作确保了TCR–CD3信号复合体的稳定组装,为后续信号传递奠定物理基础。
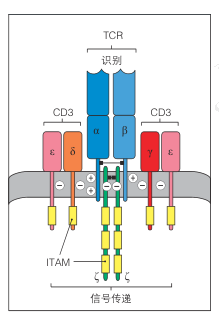
图2 TCR-CD3复合物结构示意图
当TCR识别到抗原并发生构象变化后,CD3ζ链上的ITAM序列会被Src家族激酶Lck磷酸化,从而启动下游的活化级联反应。ZAP-70(ζ-chain-associatedproteinkinase70)首先结合磷酸化的CD3ζ,继而激活并磷酸化适配蛋白LAT(linker for activation of T cells)。LAT作为信号平台,进一步招募Grb2、PLCγ1等分子,触发钙离子流入、MAPK通路和NFAT、NF-κB、AP-1等转录因子活化。最终,T细胞被全面激活,产生细胞因子、增殖并执行效应功能。
因此,TCR识别抗原不仅是一个分子识别事件,更是一个信号放大过程。从抗原肽–MHC结合到CD3复合物信号传递,再到下游转录因子驱动功能基因表达,这一完整流程确保了T细胞在面对病原或肿瘤时能迅速而精准地作出应答。
随着对TCR结构和功能认识的不断深入,TCR已从基础免疫学研究逐渐走向临床转化,其中最具代表性的应用就是TCR工程化T细胞疗法(TCR-T)。与CAR-T不同,TCR-T可以识别经MHC呈递的细胞内抗原肽,因此在肿瘤免疫治疗中具有独特优势。通过将针对特定肿瘤抗原的TCR基因导入患者的自体T细胞中,可以赋予这些细胞特异性识别并清除肿瘤的能力。早期临床试验显示,TCR-T在黑色素瘤、肉瘤和病毒相关肿瘤中已经展现了潜在疗效。
在免疫治疗靶点选择方面,TCR的特异性为靶向肿瘤新抗原提供了可能。肿瘤新抗原来源于肿瘤特异性突变,不存在于正常组织中,因而能够降低自身免疫相关副作用的风险。随着新抗原测序和生物信息学的发展,研究人员可以通过预测和验证候选新抗原,找到对应的抗原特异性TCR。这种新抗原–TCR配对的鉴定方式,为个体化治疗提供了新的策略。
同时,TCR测序在疗效评估和预测中的价值也日益受到重视。高通量测序技术能够全面描绘患者外周血或肿瘤浸润淋巴细胞中的TCR库多样性和克隆扩增情况。在免疫检查点抑制剂(ICI)治疗中,TCR克隆的扩增往往与持久的临床反应相关,而多样性下降则可能提示免疫副作用风险。因此,TCR测序不仅是科研工具,也有望成为临床决策支持和患者分层的重要方法。
TCR的研究历程充分表明,从分子结构解析到临床应用的转化路径是免疫学进步的重要驱动力。早期的结构研究揭示了TCR识别抗原的分子基础,为后续的功能研究和临床疗法开发奠定了理论框架。如今,TCR已成为免疫治疗研究的核心方向之一,不仅推动了TCR-T细胞疗法的发展,也在免疫治疗疗效预测、个体化医学和疫苗设计中展现出广阔前景。
尽管如此,TCR的特异性识别机制和交叉反应模式仍存在未解之处,不同免疫环境下的动态调控机制也尚需深入研究。要实现TCR在精准医学中的全面应用,需要结构生物学、计算免疫学和临床医学的持续合作。
总的来说,TCR的研究已经从基础免疫学走向临床转化,未来有望在肿瘤治疗、自身免疫病调控和感染性疾病防治中发挥更大作用。
参考文献
[1]Garcia KC, Degano M, Stanfield RL, et al. An αβ T cell receptor structure at 2.5 Å and its orientation in the TCR–MHC complex. Science. 1996;274(5285):209-219.
[2]Rudolph MG, Stanfield RL, Wilson IA. How TCRs bind MHCs, peptides, and coreceptors. Annu Rev Immunol. 2006;24:419-466.
[3]Zhang L, Xu X, Shi X, et al. T cell receptor signaling and cell immunotherapy. Natl Sci Open. 2024;3:20230087.
[4]Klebanoff CA, Chandran SS, Baker BM, et al. T cell receptor therapeutics: immunologic targeting of the intracellular cancer proteome. Nat Rev Drug Discov. 2023;22(12):996-1017.
[5]Huang AL, He YZ, Yang Y, et al. Exploring the potential of the TCR repertoire as a tumor biomarker. Oncol Lett. 2024;17(6):413.
记忆B细胞传统意义上被定义为生发中心(GC)B细胞的后代,表达同型转换和突变后的B细胞受体(BCRs)。然而,这不是记忆B细胞的唯一定义。我们在这里讨论小鼠的记忆B细胞,根据以下几个方面进行定义:(1)细胞表面标记;(2)多层级;(3)以T细胞依赖及GC依赖/非依赖的方式形成;(4)以不依赖T细胞的方式形成;(5)有自身免疫性疾病的小鼠模型。
未成熟B细胞表面表达BCR,这是一种与信号分子Igα/β相关的膜结合抗体。B细胞在骨髓中发育完成后,未成熟B细胞通过血液迁移到二级(外周)淋巴器官,如脾脏,在那里分化为成熟的B细胞。成人中,B细胞的发育在骨髓中进行,产生B1和B2 B细胞亚群。B1可以进一步细分为B1a和B1b,其中大部分B1a B细胞来自胎儿肝脏,而B2细胞分为滤泡(FO)和边缘区(MZ)B细胞。B1和MZ B细胞是天然抗体的来源,对非T细胞依赖(Ti)抗原起免疫反应。血液、脾脏和淋巴结中的主要亚群是FO B细胞,主要对T细胞依赖(Td)抗原做出反应。在B细胞被激活后,它们可以分化为记忆细胞和/或分泌抗体的浆细胞。
经过激活后,FO B细胞与滤泡树突状细胞(FDCs)和滤泡T辅助细胞(TFH)一起形成生发中心(GC),这是位于B细胞滤泡内的次级结构。FDCs以免疫复合物的形式捕获并保留抗原在其表面,TFH细胞被发现通过同源相互作用为B细胞提供分化信号。
GCs还支持BCR修饰,即类别转换重组(CSR)和体细胞超突变(SHM),这些过程需要激活诱导脱氨酶(AID)。GC可分为两个区,一个是B细胞进行克隆性扩增的暗区;一个是B细胞根据其与FDCs和T辅助细胞相互作用的能力进行选择的明区。当B细胞离开GCs时,它们分化为记忆B细胞或产生抗体的浆细胞,由于SHM和/或CSR导致效应功能的改变,表达的BCRs可能会经历亲和力成熟。
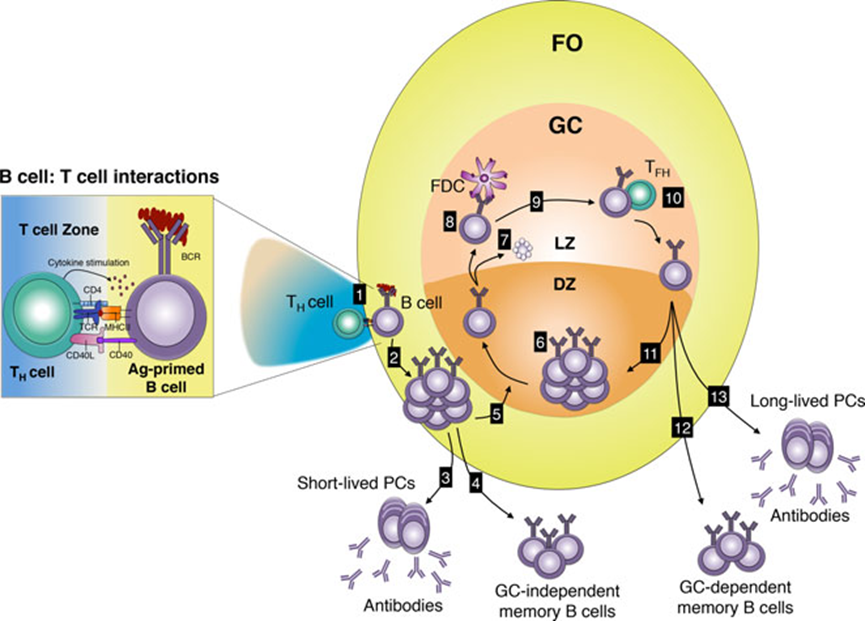
图1 记忆B细胞形成途径示意图
在人体中,记忆B细胞的比例远远高于小鼠,至少无病原体条件下的。并且人体记忆B细胞的主要特征是表达CD27,这是一种有抗原经历细胞的标志。在人类的CD27+B细胞中,存在着IgM和经历过SHM的同型转换的细胞。此外,缺乏CD27表达的记忆B细胞也有报道。有观察显示,CD27并不是小鼠记忆B细胞的适当标志物,并且由于记忆B细胞的数量少,在技术上难以仔细研究它们。为了避免这个问题,许多研究都依赖于使用杂交瘤和表达特定抗体的H链,单独或与确定的L链结合的转基因小鼠(TG),产生的B细胞可以高频率表达具有预先确定的抗原特异性的BCR。将此类结构体引入Ig的H(和L)链位点,也可以实现CSR,因此,研究可以表达同型转换抗原特异性BCRs的B细胞是可能的。
传统定义下,记忆B细胞为表达同型转换和大量突变BCRs的GC B细胞的后代。因此,对记忆B细胞的研究是针对表达同型转换抗原特异性BCR的B细胞,这些B细胞在GC反应停止后很久还存在。然而,最近在追踪记忆B细胞方面的进展使彻底研究这些细胞的性质成为可能,甚至是在不使用TG BCR的情况下。这揭示了记忆B细胞池在其生成、分化、功能、表型标记以及其BCR的SHM和/或同型转换水平方面具有未曾预见的异质性。此外,有证据表明还有一些途径可以形成记忆B细胞,这是一条Td但与GC无关的途径。不仅如此,甚至针对T细胞无关抗原,记忆B细胞也能形成。
下面根据以下几个方面讨论各种记忆B细胞群的定义:(1)细胞表面标记;(2)多层级;(3)以T细胞依赖性以及GC依赖性或非依赖性的方式形成;(4)以不依赖T细胞的方式形成;(5)有自身免疫性疾病的小鼠模型。
使用抗原特异性记忆B细胞的相对数量较高的TG小鼠模型,通过对细胞表面标志物进行研究,使记忆B细胞的定义得以确定,并且这在非TG小鼠中得到了证实。在这个系统中,B细胞表达一个明确的H链,与内源性λ1 L链结合,形成针对半抗原4-羟基-3-硝基苯乙酰 (NP)的特异性BCR。在这项研究中,用NP与鸡γ球蛋白(CGG)进行免疫后,刺激因子CD80被确定为记忆B细胞的标志物。可以与NP结合的IgM和IgG记忆B细胞被发现,并且其中60%以上表达CD80,其中大多数(70%)经过SHM。这也意味着在同型转换的记忆B细胞中,存在着表达非突变BCR的细胞,因此与传统的记忆B细胞的观点相悖。
后来CD80结合CD73和PD-L2两种标志物,区分了至少五种针对Td抗原NP-CGG免疫反应的记忆B细胞的亚群。由于在这五个亚群中都检测到了IgM和同型转换的细胞,说明亚群分类与同种型的表达无法关联。这些数据表明,记忆B细胞群的多样性是相当大的,存在一个谱系。这个谱系将由原生的记忆B细胞和其他更多的类记忆细胞组成,前者BCR不常进行同型转换且很少发生突变,而后者表达多种特异性标志物,更频繁地进行同型转换产生BCR且高突变含量高。

图2 B细胞的五种亚群的区分
在另一个模型系统中,使用黄色荧光蛋白(YFP)标记表达AID的细胞。其假设是,SHM和CSR所需的AID在GC反应期间被激活,因此YFP不仅会标记GC B细胞,也会标记它们的后代。这个模型允许长时间追踪YFP阳性细胞针对一种微粒状Td抗原的羊红血球细胞(SRBC)或可溶性Td抗原的NP-CGG所产生的免疫反应。通过这种方法,发现在SRBC免疫后长达8-12个月,都能检测到IgM和IgG记忆B细胞,而经过NP-CGG免疫后,只在3-4个月内可以检测到这些细胞,这表明对微粒抗原的反应产生的记忆会更持久,抗原的性质对记忆B细胞反应的持续时间很重要。此外,IgM记忆B细胞也有会继续分化。
对四个不同的YFP阳性记忆B细胞亚群以细胞表面标志物的方式进行区分。这些细胞可以根据IgM和IgG表达情况,以及它们是否与花生凝集素(PNA)结合进行划分。尽管所有的亚群都显示出了SHM的迹象,但无论哪种亚型,PNA阳性部分的频率都较高,并随时间而变化。此外,PNA阳性和PNA阴性的部分都是CD73和CD80阳性,只是它们的Fas(CD95)表达水平不同。记忆B细胞上CD73和CD80的表达情况与上文(1)讨论的记忆B细胞标志物一致。PNA和Fas也是GC B细胞的标志物,与此相一致的是,在SRBC免疫后的8个月内都可以检测到类似GC的结构。PNA+细胞的存在和GC反应开启了记忆B细胞再循环的可能性。事实上,IgM和IgG记忆亚群的过继转移显示,前者产生了GCs,而后者则分化为浆细胞,这也表明了记忆B细胞亚群的不同功能。由于AID的表达也可以发生在GC结构之外,YFP的阳性可能不是那些通过GC的细胞所独有的。尽管如此,这些数据与以前所认识到的更具可塑性和异质性的记忆B细胞反应相一致。基于这些结果,有人提出,B细胞记忆出现在多个层级,并具有不同的功能。
Ti B细胞反应基于抗原类型的区别可分为两种Ti-1和Ti-2。Ti-1抗原,例如细菌脂多糖(LPS),可以不考虑抗原的特异性而直接诱导B细胞的激活,它们还通过Toll样受体为B细胞提供第二个信号。Ti-2抗原,例如,肺炎球菌多糖或抗原2,4-二硝基苯偶联右旋糖酐(DNP-DE),都是高度重复的结构,可以交叉连接足够数量的BCR以完全激活抗原特异性B细胞。Ti-1抗原可以激活未成熟和成熟B细胞,而Ti-2抗原只激活成熟B细胞。Ti-2 B细胞反应主要由B1和MZ B细胞执行,并定位在滤泡外病灶。
多年来,人们认为针对Ti抗原的免疫反应不可能产生免疫记忆。早期的研究表明,初次免疫后,再次接触DNP-DE,产生的抗DNP抗体效果很差。然而,这种无反应性并不是由于缺乏抗原特异性的记忆B细胞,而是因为产生了抑制B细胞激活的半抗原特异性抗体。为了证明这一点,将DNP-DE诱导的脾脏细胞过继转移到已辐照的受体上,然后再进行刺激,结果是IgM反应增强。最近,有研究表明,B1b细胞在对Ti抗原的反应中会产生记忆B细胞,另外,B1a细胞似乎也会产生类似记忆细胞的特征。与Td B记忆细胞相比,Ti记忆B细胞在某些标志物方面出现了不同的表型。
小鼠模型中存在自身抗体会产生致病性,导致一些免疫性疾病,如系统性红斑狼疮(SLE),I型糖尿病和类风湿性关节炎(RA)。然而,产生的自身抗体本身并不一定会诱发自身免疫性疾病,相反,这些疾病的复杂的病理表现是由多个基因组合控制的。这些模型中的抗体可能来自于自体反应的记忆B细胞,尽管由于缺乏可靠的表面标记,这些细胞很难被定义和跟踪,因此大多数研究都依赖于‘传统的’记忆B细胞表型以及杂交瘤的建立。有证据表明,记忆B细胞和自身抗体都是在次级淋巴器官、受影响的局部器官中、GCs中以及在滤泡外的聚集物中形成的。
自身免疫性风湿病最常用的标志物之一是类风湿因子(RF),一种针对IgG的Fc部分的自身抗体。例如存在于易患狼疮的MRL/lpr小鼠中的自身抗体。这些自身抗体经历了CSR和SHM,而且RF特异性B细胞反应与外源性抗原引起的记忆B细胞反应非常相似。通过一个模型系统显示,具有针对RF(AM 14)特异性BCR的TG B细胞是以T 细胞依赖的方式被激活的,这发生在脾脏的T细胞区和红髓的边界,而不是在GCs中。与上面讨论的Td记忆B细胞相似,自身反应性的AM 14 B细胞可以进一步发展为CD73阳性的记忆B细胞,也可以发展为生命周期短的浆细胞。
B细胞在GCs中的存活取决于各种因素,包括细胞死亡受体Fas。该受体可以消除GC中的非特异性和自发性的B细胞;因此,如果Fas或FasL信号通路被破坏,自体反应记忆B细胞和浆细胞的生存和产生就会被允许。相反,在选择抗原特异性非自体反应的B细胞的过程中,其他逃逸信号确保了对Fas介导的细胞凋亡的抵抗。事实上,MRL/lpr小鼠出现的类似系统性红斑狼疮的综合征,包括肾小球肾炎、多动脉炎、关节炎和咽喉炎,是由于缺陷的Fas基因(lpr)与其它身份不明的突变基因的结合。
已知GCs的自发形成发生在自身免疫性小鼠的次级淋巴器官中,如自发性糖尿病NOD小鼠和狼疮模型 (MRL/lpr, PN, NZB, NZB/W, B6/lpr and BXSB 雄性小鼠),在没有免疫或感染的情况下,这些1至2个月年龄的小鼠已经发生了。基于用抗CD40配体抗体治疗后GCs的消退,在自身免疫中形成的GCs和由免疫引起的GCs都是T细胞依赖性的。已知在自身免疫环境中GCs的自发形成也发生在受影响的器官中,例如在糖尿病NOD小鼠的胰岛和胶原蛋白诱导的关节炎(RA最常用的小鼠模型)的滑膜组织中。在糖尿病NOD小鼠中,胰岛的GCs与次级淋巴器官中的GCs非常相似,因为它们都含有FDCs和T细胞,存在B细胞上调AID以及表达体细胞突变的寡克隆BCR组合。此外,GC B细胞可以在原地分化为可以产生针对胰岛素的抗体的浆细胞,也很可能分化为记忆B细胞。在患有胶原蛋白引起的关节炎的小鼠的关节中,大约50%的B细胞表达了GC标志物,如GL7和Fas,而同一小鼠脾脏中GC B细胞的相应比例约为10%。因此,很可能小鼠自身的GCs有助于记忆B细胞和浆细胞的产生。
综上,用于研究小鼠记忆B细胞的模型系统对实验结果很重要,因为它们在初次(和再次)免疫反应的持续时间、GCs的持久性和记忆亚群方面存在差异。实验结果还取决于抗原的剂量和类型、免疫的时间间隔以及用于定义记忆B细胞的标记物。然而,之前的数据表明,记忆B细胞的形成有两种途径,一种是是GC依赖性的,另一种不依赖。这两种途径都需要T细胞的帮助,并产生IgM和同型转换的记忆B细胞。如所讨论的转换和非转换记忆B细胞的五个亚群,也可能符合这两种途径,也许代表免疫反应的不同阶段。沿着其中一条途径,记忆B细胞将被生成并表达未变异的抗体,保护宿主免受各种入侵者变异体的伤害,而另一种途径将产生记忆性B细胞,它能以高亲和力、变异和同型转换的抗体迅速作出反应,并提供对同一抗原的再次变异的防御。Ti抗原也可以在同型转换和非转换B细胞中产生记忆反应。
在自身免疫条件下,自体免疫反应最初可能与那些外源性抗原所驱动的途径一样。然而,由于致病的自身抗体主要是突变的和同型转换的,这可能表明自身抗原的持续存在使反应偏向于长期的GCs和永久产生GC依赖性记忆B细胞和自身抗体产生的浆细胞。
决定B细胞命运的机制,也就是说,是什么使细胞走早期记忆B细胞而不是GC B细胞的途径,以及什么使GC B细胞分化为记忆B细胞而不是浆细胞,目前仍不清楚。到底是一个信号,还是几个信号将B细胞引向某条道路,目前还不完全清楚,也许是在内在和外部信号的影响下,例如抗体反馈机制。或者,决定性的事件可能只是随机的,例如,细胞分裂时抗原的不平等分配,产生的后代具有不同的呈现抗原和竞争T辅助细胞的能力。
参考文献
Bergmann B, Grimsholm O, Thorarinsdottir K, et al. Memory B Cells in Mouse Models[J]. Scandinavian Journal of Immunology, 2013, 78(2).






南京德泰生物工程有限公司 Nanjing Detai Bioengineering Co.,Ltd. ©2026 All Rights Reserved
 苏公网安备32011202001300
苏公网安备32011202001300