
近年来,抗体药物凭借其高靶向特异性、低免疫原性和长效药代动力学特性,在多种疾病治疗领域获得广泛应用,获批上市数量显著增加。得益于现代生物技术的进步,抗体发现平台日益成熟,具备特定功能的生物大分子持续涌现。然而,抗体药物研发仍面临较高的失败率,导致最终获批药物有限。其核心挑战在于分子固有的易聚集性、稳定性不足和溶解性差等特性,这些缺陷为后续的生产工艺、制剂开发、储运环节带来显著困难,阻碍了药物成功开发。因此,在早期分子筛选中,亟需运用体外方法评估分子的均一性、稳定性、溶解度和结合特异性,筛选出优势候选分子,并识别其潜在风险。这一过程——成药性评价——旨在降低因分子自身不良属性导致的后期研发失败风险。
成药性评价主要通过一系列高通量的技术手段,对抗体的一级结构、翻译后修饰、聚体和片段、电荷异质性、疏水性、构象稳定性、胶体稳定性、溶解度、黏度、蛋白质-蛋白质相互作用、非特异性相互作用、表位竞争和亲和力等方面进行多维度的分析,筛选出最佳的候选分子,确保在后续的开发中具有较为稳定的性质,从源头上保证产品的质量。
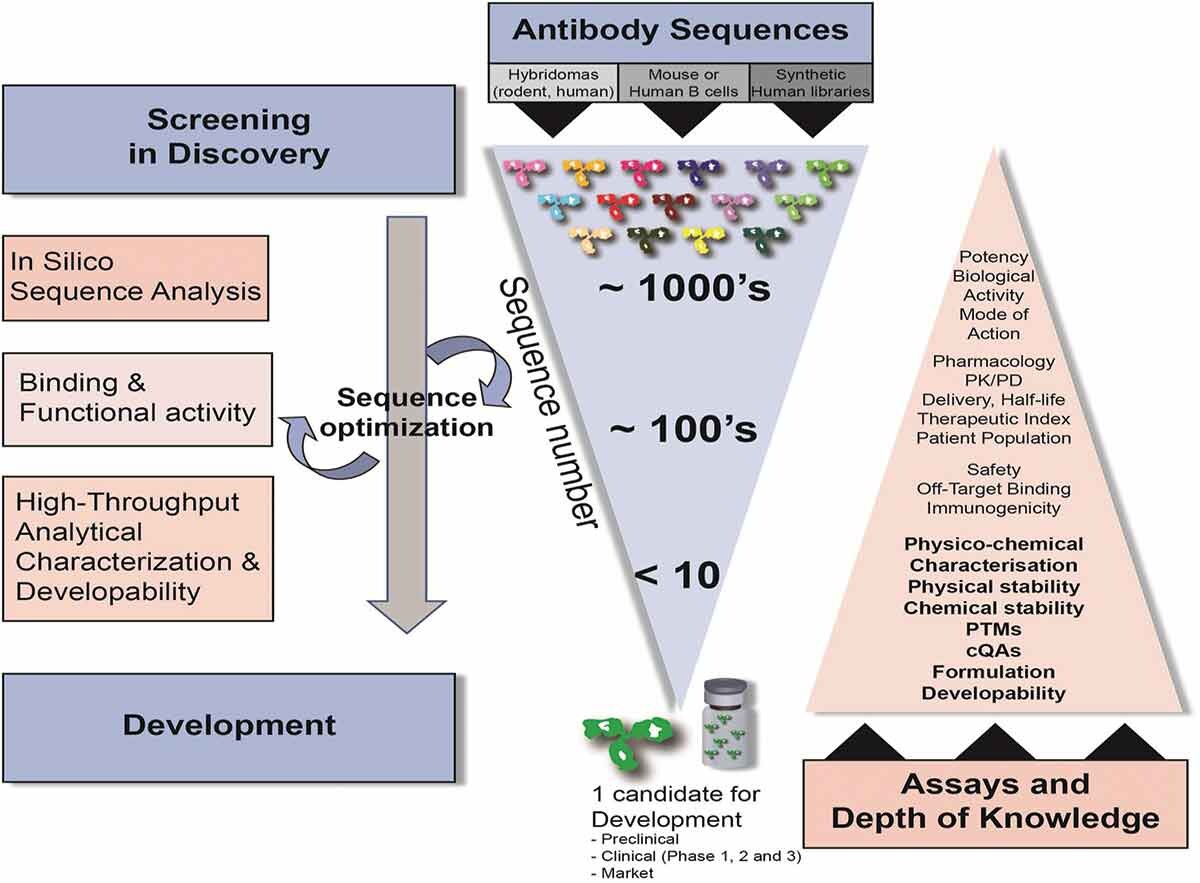
传统上,抗体药物发现阶段的筛选过程被认为是漏斗型筛选,通过杂交瘤技术、展示技术(噬菌体展示、细胞表面展示、无细胞分子展示技术)、人源抗体转基因小鼠和人杂交瘤技术、展示技术(单B细胞测序等技术,获得数量庞大的候选序列,以与目标种属蛋白特异性结合、功能活性等评价方法作为主要评价策略进行多轮筛选。但目前越来越多的研究者将成药性评价提前至抗体药物发现阶段,有利于尽早评估和解决候选药物的可开发性问题,最终获得一个均一、稳定、安全、有效和可放大生产的候选序列,提高药物开发的成功率和效率。
抗体药物早期发现阶段的成药性筛选流程划分为成药性初期评价、成药性筛选和成药性评估3个阶段。成药性初期评价和筛选阶段,样品种类多、数量大,综合时间进度和有限的资源,采用倒漏斗型筛选策略,选择少量且关键的质量属性进行评价,同时配合高通量技术加快评估流程;对中后期筛选出的分子,应进行多维度且较为全面的质量属性的表征;对最终筛选出的分子,应增加加压实验等研究,以获得更为全面的分子属性。值得注意的是,在抗体药物发现阶段,成药性筛选评估没有绝对的标准对候选分子进行剔除,而是对候选分子综合评估后,打分排序(如风险等级按高、中、低排序),帮助及时了解候选药物的分子属性和潜在风险因素,掌握控制要点,便于后续工艺和制剂的开发。
非人源的抗体候选序列(如杂交瘤筛选技术)获得后,需先构建为嵌合抗体,即对候选序列进行恒定区人源IgG1或IgG4代替,使用哺乳动物细胞系(人源胚胎肾细胞、中国仓鼠卵巢细胞等)表达;对于筛选获得的人源序列,可以直接进行质粒构建和表达;对于抗原结合片段库(Fab)、单链抗体可变区基因片段库(scFv)和单域抗体库(dAb)筛选得到的序列,可以制备成IgG-like的形式,也可以选择合适的表达体系进行单独表达。此阶段的表达方式一般为瞬转表达,有利于快速获得小体系样本,经高通量设备纯化,得到具有一定纯度的样品。
此阶段是以活性筛选策略为主,包括结合活性[酶联免疫吸附技术(ELISA)、流式细胞技术(FCM)]和功能活性(天然细胞、工程细胞),还可以借助高通量分子相互作用仪,如基于表面等离子共振技术(SPR)的BiacoreTM 8K系列仪器或者基于生物膜干涉技术(BLI)的Octet® BLI系列仪器,检测与目标种属靶抗原的特异性、亲和力、结合动力学和表位竞争等。在保证项目进度的前提下,可选择关键物理化学属性进行成药性初期评价,包括表达量和纯度。纯度可通过高通量技术进行检测,如体积排阻色谱-高效液相色谱(SEC-HPLC)法、十二烷基硫酸钠-聚丙烯酰胺凝胶电泳(SDS-PAGE)法或毛细管凝胶电泳-十二烷基硫酸钠(CE-SDS)法等。瞬转表达量与稳转表达量具有一定的相关性,筛选高表达量的分子,同时选择不易形成聚集或降解的分子,不仅可以降低后期生产成本,还代表着具有更优的构象稳定性和胶体稳定性,也为体外活性检测奠定基础,避免因聚集体或降解物过多导致结果失真。
筛选出的序列若用于制备双特异性抗体,应在此阶段进行组装考察,评价活性和稳定性,可以通过构建质粒表达,也可以借助体外重组技术完成。评估项目包括表达量、纯度和活性,值得注意的是:纯度检测项应根据分子及其变异体性质的差异选择合适的评价策略,如可根据电荷差异通过离子色谱-高效液相色谱(IEX-HPLC)和成像毛细管等电聚焦(iCIEF)等方法进行表征,也可根据分子量大小通过SEC-HPLC和毛细管凝胶电泳法表征。
筛选出的序列若用于制备抗体偶联药物,可在此阶段进行偶联,评价分子偶联效率和稳定性,如通过SEC-HPLC等方法检测聚体和片段的含量、通过疏水相互作用-高效液相色谱(HIC-HPLC)法或质谱法检测ADC的偶联量及偶联位点的分布、通过质谱法检测小分子脱落量和通过ELISA法或质谱法检测血浆稳定性等。
经以上步骤筛选出的分子,借助高通量计算机模拟和分析技术,对氨基酸序列、分子结构、蛋白质和蛋白质相互作用进行初步分析和预测。针对抗体中的非人源性成分,进行人源化改造,以降低在人体内出现免疫原性的风险,保留抗体互补决定区非人源氨基酸残基,其余均替换为人抗体的相应部分,或改造暴露在抗体表面的骨架区的非人源氨基酸残基;对于全人源抗体库筛选出的人源抗体,需进行亲和力成熟等改造。不同的突变改造可能会影响分子稳定性,应综合考虑平衡亲和力、免疫原性和分子稳定性;对于其他潜在的一些问题,如等电点、脱酰胺、异构化、暴露的色氨酸和蛋氨酸、未配对半胱氨酸、糖基化或糖化等也应给予关注和改造,如等电点过高可能会影响半衰期,等电点过低在常规的工艺过程中易产生聚集;互补决定区的脱酰胺、异构化导致分子降解或者活性降低;氧化可能会影响活性,也可能引发聚集体的产生;可变区的糖基化或糖化位点,可能会影响活性和稳定性等等。
改造后的分子通过轻重链的组合配对,产生一系列的变体,数量一般在100个左右,通过哺乳动物细胞瞬转表达和高通量纯化(依据活性实验需求进行内毒素控制),评价表达量、纯度和生物学活性,去除极差分子后,开展成药性筛选评价。
成药性筛选阶段的目标为对各分子的固有属性进行评估和排序,如电荷变异体含量、疏水性、热稳定性、不同粒径聚集体、自相互作用、非特异性相互作用、与不同种属抗原的亲和力、表位差异、Fc功能等。最终挑选出具有较低异质性、较高稳定性、较长半衰期、高产量和可工艺放大的抗体,筛选出的分子数量一般小于5个。本阶段成药性筛选所用到的检测技术,应具备样品用量少、通量高、分析快速等特点。若候选分子具有特殊属性,可在此阶段增加初步的稳定性评估,以确保样品在检测、存储、动物给药过程中的稳定,如室温稳定性、2−8℃稳定性、冻融稳定性等。若候选分子计划用于制备高浓度剂型,可以增加溶解度预测实验,随着蛋白浓度的升高,相较于低浓度溶液,蛋白与蛋白之间的作用力增强,但传统的溶解度测定方法对蛋白消耗量大,故可选用在较低浓度中预测溶解度的方法。
对于最终筛选出的几个候选分子,进行进一步的可开发性评估。表达阶段扩大瞬转体系,纯化阶段增加精纯步骤,并严格控制内毒素含量,为早期的动物体内药代动力学(pharmacokinetics, PK)、药效动力学(pharmacodynamics, PD)以及毒理研究储备样品。成药性筛选阶段进行的成药性表征方法应再次检测和确认,另外还应进行加压实验的考察,有助于快速地暴露分子的风险位点及变化趋势,考察对抗体特异性和功能活性的影响程度,考察分子的耐受性和可及性,评估敏感条件,预测长期稳定性,如设置40℃高温稳定性,纳米抗体可进行100℃高温稳定性考察。通过翻译后修饰、聚集体和降解物含量、酸碱变异体含量、ADC中小分子脱落量的变化,发现分子潜在的翻译后修饰位点、降解位点及变化趋势;还可以设置强酸、强碱、氧化和光照等加压实验;对于高浓度制剂的开发,在前期溶解度预测实验中,由于高的稀释因子,难以准确推导高浓度制剂真实的溶解度和黏度,故在此阶段,可以开展目标浓度的浓缩和过浓缩,使用常用缓冲体系,添加尽可能少的制剂辅料,观察药物在浓缩过程中的特性,包括但不限于蛋白含量、聚集体和降解物的含量、粒径的尺寸分布和分散度、黏度、热稳定性、储存稳定性以及在高浓度下与血清的相融性等。最佳配方条件的探索,则应在后期制剂开发中进行。
成药性评价作为一种在药物发现阶段被广泛认可的候选分子筛选方式,已经成为抗体药物研发过程中的关键一环。不同成药性筛选阶段,选择符合筛选目的的评估策略,不仅能理解分子目标属性,还能大幅减少筛选时间,降低冗余数据堆砌,最终加快筛选流程和提高研发成功率。
参考文献
姚江宁, 律璎桐, 张迎珺, 张正平, 徐同杰. 抗体成药性筛选流程及评价策略[J]. 生物工程学报, 2024, 40(2): 507-516






南京德泰生物工程有限公司 Nanjing Detai Bioengineering Co.,Ltd. ©2026 All Rights Reserved
 苏公网安备32011202001300
苏公网安备32011202001300