
杂交瘤技术是一种将B淋巴细胞与骨髓瘤细胞融合生产单克隆抗体的传统方法。该技术通过将动物脾细胞与骨髓瘤细胞融合,产生永生化杂交瘤细胞系,然后筛选其上清液中的抗原特异性克隆,并进一步循环亚克隆以产生严格的单克隆抗体。
利用杂交瘤技术制备单克隆抗体通常操作流程为:抗原制备、抗原免疫动物、杂交瘤细胞制备、融合细胞的筛选、培养杂交瘤细胞制备抗体。(详细实验流程:杂交瘤单克隆抗体制备SOP)在单克隆抗体制备过程中,常常会遇到细胞污染,细胞融合后不生长、亚克隆后的细胞株不分泌抗体以及细胞难以克隆等问题,本文将详细分析以上常见问题的产生原因以及解决该问题的方法。
一旦确认有分泌抗体的杂交瘤细胞,就应尽快进行克隆化。克隆化的目的是为了获得单一细胞系的群体,反复克隆化后可获得稳定的杂交瘤细胞株。杂交瘤细胞若难以克隆化,可以尝试以下解决方法:
蛋白质磷酸化(phosphorylation)对于调控细胞功能而言是相当重要的,细胞生长、细胞增生、细胞中的讯号传递等现象都与蛋白质磷酸化息息相关,因此蛋白质磷酸化也是细胞生物学领域研究非常重要的一环。
Western blot是研究中最常使用来评估蛋白质磷酸化状态的实验技术,但想要把蛋白质磷酸化的Western Blot实验做好却往往是研究人员最大的梦魇。

因此,在这里和大家分享此类型实验时应该要注意的几个事项与诀窍:
在实验前需先查阅文献或通过预实验检测该样本是否表达目的蛋白,可以通过Uniprot、PhosphoSitePlus等数据库查询蛋白的磷酸化情况。正常情况下样本的磷酸化水平过低或不表达,可通过物理或化学方法进行一定的刺激和诱导,刺激条件各不相同,刺激的时间和强度也需要查阅文献和进行预实验来确定。若表达量偏低或根本检测不到则建议可以将样品以蛋白质组学的方式进行检测。若该样本根本就不表达目的蛋白,那就需要重新设计实验方案。在实验进行时,需要设置适当的阳性和阴性对照,可进一步确定抗体检测到的信号的特异性。
蛋白的磷酸化是一个非常迅速的反应,所以样品要新鲜,提取磷酸化蛋白的裂解液最好是新鲜配制,避免反复冻融;样品处理最好在冰上操作,操作时间尽量短;用PBS洗涤细胞时,PBS一定要4℃预冷;如果样品来源是组织,提取蛋白时采用液氮研磨的方法,研磨器具最好提前预冷,保持低温的状态。
提取样品的过程要迅速,组织或细胞裂解时会释放出大量内源性蛋白磷酸酶,它可以催化磷酸化蛋白的去磷酸化,导致不同于正常生理状态的差异。裂解液中一定要有蛋白酶抑制剂和磷酸酶抑制剂,否则即使条带出来也会很浅,结果可信度不高。除了内源性的蛋白磷酸酶外,也存在外源性的磷酸脢,通常是实验过程中的污染所造成的,因此在样品制备的过程当中请务必使用不含磷酸脢的试剂,而值得特别注意的是样品中或是实验环境中的细菌也会分泌磷酸脢,要特别留意。另外,还要注意检测蛋白磷酸化位点是什么氨基酸,如果是酪氨酸还要加1µmol/L的原钒酸钠,上样前不要煮沸,煮沸可能会破坏其磷酸化位点。提取的磷酸化蛋白需要妥善保存,收集完成后尽快蛋白变性并于-80℃分装保存,以保证降解程度最小。同时避免反复冻融导致磷酸基团的降解。
磷酸化蛋白检测需要设置两个内参照,一个是磷酸化蛋白对应的总蛋白,另一个是普通蛋白检测的内参比如actin和GAPDH等。普通内参作为体系的对照,保证上样量一致从而排除体系本身的影响,对于结果分析比对十分重要。
同时,总蛋白检测也是必需的,只检测磷酸化蛋白质的表达量并无法真实的说明细胞中磷酸化状况,只有将磷酸化蛋白质与总蛋白质相比,比值的升高或是降低才是有意义的。
通常好的抗体可以决定70% Western Blot的成败,所以挑选好的磷酸化抗体绝对是一个非常重要的关键。德泰生物提供磷酸化鼠/兔单克隆抗体制备服务,在设计免疫原、合成磷酸化多肽及磷酸化抗体筛选、制备技术上具有丰富的经验,最终交付的抗体将通过磷酸化多肽与非磷酸化多肽的交叉验证。
磷酸化蛋白转膜后,使用5%BSA(TBST溶解)进行封闭。避免膜封闭时间过长,因为这会掩盖抗原表位,阻止抗体结合。用洗涤时摇床的转速不能太快,洗涤的时间不能太长,洗涤过程每张膜分开洗涤。由于磷酸化蛋白表达量低,占总蛋白量的极少部分,因此抗体孵育时可加大一抗浓度后4℃过夜,保证抗体有充分的结合时间。一抗孵育完毕后回收抗体重复使用,二抗孵育室温1h。
抗体洗脱液用来去除已结合的抗体,但也会去除部分蛋白,导致蛋白信号变弱。实验过程水浴温度须准确控制,水温过高会导致较多目的蛋白被去除。抗体洗脱时膜一定要完全泡在抗体洗脱液中,将塑料膜完全浸入水中使其受热均匀,否则导致显影出来的总蛋白不均一,影响结果分析。
抗体药物偶联物(ADC)偶联肿瘤抗原特异性单克隆抗体mAb、稳定的化学linker与强效细胞毒素,将单抗的选择性与化疗药物的细胞毒性潜力相结合。这三种组分共同产生了一种强大的溶瘤剂,能够在抗体对肿瘤靶标的高亲和力的引导下,特异性地将正常耐受的细胞毒性药物递送到癌细胞(图1)。
在过去的十年中,对于ADC研究工作集中在linker和优化有效载荷上。抗体作为传递细胞毒性有效载荷的重要机制,与抗体独特结构特征相关的其他潜在影响仍在探索之中。

图1 ADC各组成部分及其在ADC设计、工程和功能中的作用。
ADC的成功开发取决于选择合适的靶抗原,这是ADC疗效的决定因素。ADC等靶向疗法利用癌细胞和正常细胞之间蛋白表达的差异来选择合适的靶抗原。目前正在开发的靶标包括HER2(即曲妥珠单抗美坦新)和CD30(即布伦妥昔单抗维多汀)。人们普遍认为,靶抗原应在肿瘤细胞表面均匀且选择性地表达,在正常组织中很少或没有表达,以限制脱靶毒性。
ADC的靶标验证必须基于靶抗原的可靠鉴定。基因组研究指出,肿瘤细胞具有复杂的异质性,存在不同细胞亚群,不同亚群具有独特的表型多样性,这些差异来源于肿瘤内和肿瘤间遗传和非遗传的异质性影响。这对靶标的选择提出了深刻的挑战。虽然ADC疗法并非要求靶抗原表达绝对均匀(异质性肿瘤可能因旁观者效应而使治疗效果提升),但最新临床开发中的ADC主要适用于具有基本一致的谱系特异性标志物的血液肿瘤适应症。
传统对靶抗原的选择主要集中在肿瘤细胞上表达部分,然而人们对靶向肿瘤微环境中存在的抗原越来越感兴趣,包括新生血管、内皮下基质和肿瘤基质内的抗原。靶向基质的ADC通过降低基质产生的生长因子的浓度导致肿瘤细胞死亡。由于所有肿瘤细胞的存活都依赖于血管因子和基质细胞生长因子,因此靶向此类组织的ADC可能具有更广泛的疗效。这种靶向策略特别有吸引力,因为与癌细胞不同,这些细胞在基因组上是稳定的,并且不太可能产生与突变相关的耐药性。
靶抗原应通过受体介导的内吞作用很好地内化,不应因内吞作用或治疗期间重复刺激而使得作用下调。为了使ADC产生临床效果,抗原表位的识别必须导致内吞作用。一般来说,内化良好、在正常组织上表达低、在肿瘤上表达高的抗原是ADC方法的首选。然而,临床试验的结果表明,健康组织中的靶抗原表达可能会导致脱靶毒性难以预测。与非偶联抗体功能不同,目前的实验证据通常表明抗原密度与ADC的功效没有直接关系。对淋巴瘤、黑色素瘤和前列腺癌的研究表明,ADC的抗原密度与治疗反应之间没有直接相关性,ADC疗效的发挥需要最低抗原表达阈值。
ADC的抗体部分的主要作用是选择性地将细胞毒性药物递送到靶细胞。因此,在设计ADC时,对抗体中负责抗原识别的Fab部分给予了很大的关注。用于开发ADC的抗体主要是全长抗体,几乎完全属于IgG类。然而,对抗体的Fc段的考虑较少。
1)对效应功能的影响
IgG抗体的Fc段包含与新生儿Fc受体(FcRn)的结合结构域,可调节血清半衰期,并识别免疫效应细胞上不同的活化和抑制性Fc受体,这些受体可影响生物利用度、组织隔离、肿瘤转运、抗原靶向和免疫功能。
完整的IgG ADC能够将补体成分和免疫效应细胞募集并激活到肿瘤部位,介导二级免疫功能,如补体依赖性细胞毒性(CDC),抗体依赖性细胞介导的细胞毒性(ADCC)和抗体依赖性细胞介导的吞噬作用(ADCP)。ADC触发免疫效应功能的能力可以提高抗肿瘤活性,也可以通过循环中的免疫细胞隔离ADC并影响ADC在肿瘤部位的定位和靶细胞内化,或者被免疫细胞内化导致脱靶毒性。研究表明,裸抗与相应的ADC之间具有相似的抗体介导的效应功能。相反,也有研究确定Fc受体会引起ADC治疗副作用。T-DM1已被证明可通过与FcγRIIa结合,在体内被巨核细胞内化,这与T-DM1诱导的血小板减少症的发展有关。除了FcγRIIa结合之外,还有其他机制,例如巨胞饮作用,这就可以解释巨核细胞非受体/非靶点介导的摄取而引起的血小板减少症。
综上所述,根据肿瘤的类型、抗原的表达以及抗体对抗原的亲和力,设计具有Fc介导功能的ADC是合适的,可以通过选择合适的IgG亚星或设计Fc区域来优化ADC。
2)抗体亚型的影响
IgG有4个亚型,到目前为止除IgG3外的所有亚型都已用于开发目前正在临床试验中的ADC。
IgG1是ADC设计中最常用的亚型。它具有与IgG2和IgG4相当的血清稳定性(21天),但具有更强的补体固定能力和更高的亲和力,激活单核细胞和巨噬细胞(FcγRI、FcγRIIa、FcγRIIIa)和NK细胞(FcγRIIIa)等效应细胞上表达的FcγR。因此,该亚型具有激活免疫系统并触发CDC,ADCC和ADCP的卓越能力。
IgG3具有锚定补体、结合激活FcγR的优越能力,但与其他亚型相比,IgG3在血清中的半衰期较短,其长铰链区易于被水解以及潜在免疫原性,迄今为止IgG3亚型的抗体在ADC开发中尚未被采用。
IgG2、IgG4锚定补体的能力较弱,且对FcγR的亲和力较低。当不需要激活免疫系统时,IgG2和IgG4可用于治疗性抗体的设计。IgG2有四个二硫键,而IgG1和IgG4只有两个,因此它似乎更适合搭载更高DAR的马来酰亚胺接头。IgG2也能够形成二聚体,但二聚化对治疗效果的影响需要更深入的研究。IgG2 ADC,如AGS-16M8F(抗ENPP3 IgG2-MMAF),目前正在临床试验中进行评估。
IgG4是具有非常不寻常特征的亚型,例如能够与其他IgG4抗体进行Fab臂交换(FAE),因此可以导致双特异性和功能单价形式,抗靶标功能降低。因此,通常引入特定的单点突变可以稳定抗体并预防FAE。尽管与IgG1相比,IgG4激活FcγR的亲和力较低,但对FcγRI的亲和力足以进行功能激活,并且其去岩藻糖基化形式能够结合FcγRIIIa。因此,在设计IgG4的ADC时,需要考虑免疫细胞活化。FDA在2000年批准的第一个ADC就是使用IgG4亚型抗体(Gemtuzumab ozogamicin, anti-CD33 IgG4-Calicheamicin),该药由于批准后的研究显示患者生存率没有明显改善,于2010年自愿退出市场。然而,这种ADC今年已被重新批准用于治疗急性髓性白血病。目前更多的IgG4 ADC正在临床试验评估中。
3)Fc段的工程策略
调节ADC参与免疫系统反应能力的另一种方法是设计其Fc的结构域,最广泛使用的方法是抗体糖工程。
如果选择的抗体亚型是IgG1,可以在Fc段引入单点突变或突变组合,以增强或减弱IgG1与FcγR或补体(C1q)的结合,从而增强或减弱ADCC、ADCP或CDC。举个例子,I期临床试验的MEDI4276(anti-HER2 IgG1-tubulysin analogue)通过三个单点突变(E234F,S239C和S442C)以减少FcγR结合,目的是最大限度地减少T-DM1引起的血小板减少症。
Fc改造也可用于改善ADC的药代动力学。人源化IgG1 MEDI-524-YTE,具有三个单点突变(M252Y,S254T和T256E),以增强IgG1与FcRn的结合。与野生型抗体相比,MEDI-524-YTE在食蟹猴中的血清半衰期增加了四倍。值得研究的是,增加FcRn结合和延长半衰期是否会导致ADC的活性增加和毒性降低。
4)基于生物分布考虑的ADC设计
独立的小分子治疗剂可以在体内广泛分布。相比之下,ADC通常保留其抗体成分的药代动力学特性,因此表现出相对较低的清除率和更长的半衰期。
尽管对治疗性抗体进行了优化,且抗体对肿瘤的亲和力高于正常组织,但到达肿瘤的抗体量仅为给药量的一小部分(约1-2%)。抗体片段(如Diabody)比IgG小得多(约50 kDa),因此具有卓越的组织穿透能力。然而,由于它们的尺寸较小且缺乏通常与FcRn结合的Fc部分,因此抗体的清除速度比全长IgG快得多。研发中的抗CD30-Diabodies-药物偶联物已经显示出很高的抗肿瘤活性,但是,在ADC设计中使用的Diabodies需要进一步研究和优化,以便在最佳组织渗透和低清除率之间取得平衡。
在过去的十年中,ADC研究工作集中在linker和优化细胞毒性有效载荷上。ADC的抗体支架为肿瘤相关抗原提供所需的特异性,将细胞毒性药物转运到癌症组织。然而,如何筛选适合抗体及其结构特征的潜在临床效果仍然没有得到很好的探索,甚至可能没有得到充分利用。
在设计下一代更有效、特异性更强的ADC时,我们可以考虑通过抗体部分的结构来进行优化。选择合适偶联方法可以保持抗体稳定性,并且在需要时募集和优化免疫细胞的参与。将来,具有特定抗体同种型的Fc结构或改造后的Fc结构可能会提供更好的生物分布控制,并改善免疫细胞的参与和激活。以抗体结构属性为依据的新颖设计方法将为下一代ADC的成功做出贡献。
参考文献
Ricarda M. Hoffmann, Ben G. T. Coumbe, Debra H. Josephs, et al. (2018) Antibody structure and engineering considerations for the design and function of Antibody Drug Conjugates (ADCs), OncoImmunology, 7:3, DOI: 10.1080/2162402X.2017.1395127
脂质纳米颗粒(LNP)作为COVID-19 mRNA疫苗的重要组成部分,在有效保护mRNA组分及将mRNA转运到细胞发挥着关键作用。除了制药行业,LNP的应用也已扩展到其他领域,如医学成像、化妆品、营养、农业以及纳米反应堆等其他创新领域。
脂质体是LNP的早期版本,是一种极其通用的纳米载体平台,它们可以运输疏水或亲水分子,包括小分子,蛋白质和核酸。事实上,脂质体是最早成功从概念走向临床应用的纳米药物递送平台。许多脂质体药物制剂已被批准并成功应用于医疗实践。
下一代LNP包括固体脂质纳米颗粒(solid lipid nanoparticles, SLN)、纳米结构脂质载体(nanostructured lipid carriers, NLC)和阳离子脂质核酸复合物,表现出更复杂的内部结构和增强的物理稳定性。LNP能够控制体内药物递送的位置和时间,可用于治疗各种疾病。越来越多的科学家正在超越传统的生物制药,转向更复杂和更专业的疗法,可以在基因水平上对抗疾病。
脂质体(liposome)是具有封闭的脂质双层的囊泡结构,可以在水中自发形成,最早于20世纪60年代提出(图1-A)。脂质纳米颗粒一词在1990年左右开始使用。由于脂质体在大多数情况下是纳米级的,因此它们理所当然地被认为是脂质纳米颗粒的最早一代。

图1 常见脂质体类型
(A)脂质体
(B)包封疏水和亲水药物的脂质体
(C)用靶向配体功能化的免疫脂质体
(D)用惰性聚合物(如PEG)功能化的空间稳定的脂质体
脂质体作为药物递送系统的潜力随后立即就被认识到。已知超过40%的用于癌症治疗的小分子药物在水中表现出低溶解度,因此能够包封这些药物并提高其水溶性的药物输送系统的好处显而易见。脂质体是最早成功从概念到临床应用的纳米药物递送平台,拥有多项获批的药物制剂。例如,最早批准的脂质体药物是Doxil,是一种抗肿瘤剂阿霉素的脂质纳米颗粒制剂,用于治疗卵巢癌。另一种脂质体药物Epaxal是用作肝炎疫苗的蛋白质原的脂质纳米颗粒制剂。许多其他脂质体制剂已被批准用作药物和疫苗,提供抗癌、抗炎、抗生素、抗真菌、麻醉剂以及其他药物和基因疗法。
脂质体由一个或多个脂质双层组成,大小在20∼1000 nm之间。亲水性药物可以封闭在脂质体的水性内部,而疏水性药物可以包留在脂质双层的烃链区域中(图1-B),使脂质体成为多功能药物递送平台。脂质体按照脂质双分子层数和粒径不同,可以分为直径为20~100nm的小单层囊泡(SUV),直径为100~1000nm的大单层囊泡(LUV),直径为>1000nm的巨型单层囊泡(GUV),以及直径为>500nm的多层囊泡(MLV),其内部为同心洋葱状多层结构(图2)。药物输送系统主要使用SUV和较小的MLV,而GUV主要用作细胞模型。
粒径大小是决定脂质体药物包封和循环半衰期的关键参数,较小的脂质体有更多的机会逃脱吞噬细胞的摄取。普遍认为,用于制药目的的脂质体颗粒,特别是用于肠胃外给药的颗粒,需要≤100nm。纳米颗粒的尺寸可以使用多种技术进行测量,例如动态光散射、尺寸排阻色谱、核磁共振波谱和显微镜。LNP的粒度分布可以通过挤出法、超声法和均质等制造方法进行控制。最近,微流体方法已成功用于LNP制造和尺寸控制。

图2 不同粒径和脂质双分子层数的脂质体
随着疾病细胞层面遗传学机制研究的进展,使得针对人类许多疾病基因进行靶向治疗成为可能。核酸疗法包括DNA药物疗法和RNA药物疗法。然而,核酸疗法的发展受到细胞递送困难的阻碍。核酸的负电荷和亲水性阻碍了它们在质膜上的被动扩散。此外,核酸与血清蛋白的结合、吞噬细胞的摄取以及内源性核酸酶的降解会干扰其有效递送。因此,核酸需要递送载体来保护它们免受降解,并将其递送到靶细胞以实现有效摄取。常用的递送载体包括病毒和非病毒载体。阳离子LNP由阳离子脂质和阴离子核酸之间的稳定复合物组成,是核酸药物使用最广泛的非病毒递送系统。
已有大量的阳离子脂质作为核酸载体被合成与测试。阳离子脂质的分子结构与天然脂质相似,差异仅存在于可电离(阳离子)头基而不是天然脂质的两性离子或阴离子头基。阳离子脂质包括具有两个烷基链或胆固醇疏水部分、带正电荷的极性头基团以及连接极性基团与疏水部分的linker。阳离子脂质仅在细胞内带正电荷,而在血液中由于pH值的变化不带电荷,所以它们比非可电离阳离子脂类的毒性小。
与带正电荷的脂质络合可稳定核酸并增强其对核酸酶降解的抵抗力,使其能够递送到所需的靶细胞。核酸通过LNP吸附到细胞表面进入细胞,然后进行内吞作用并将核酸释放到细胞中。由于细胞膜通常带负电荷,而用于核酸递送的纳米颗粒脂质带正电荷,因此静电作用促进了LNP对细胞膜的吸附和与细胞膜的融合,驱动了膜融合和内吞作用。一旦核酸进入细胞,核酸需要从其与阳离子脂质的复合物中释放出来。细胞膜的阴离子脂质可能通过中和阳离子脂质载体的电荷,破坏脂质载体和核酸之间的静电相互作用来帮助从LNP释放核酸。阴离子脂质与阳离子脂质的结合也会破坏纳米颗粒结构,导致非层状结构的形成。阳离子脂质载体在递送核酸方面的功效被认为与其促进非层状脂质相形成的能力相关。短寿命的非层状结构被认为介导膜融合过程;在膜融合中形成的中间体与在层状-非层状相变中形成的中间体相似。

图3 脂质纳米颗粒核酸载体的推荐结构
(A)核酸位于反向脂质胶束组织中
(B)核酸插层在脂质双层之间
虽然脂质体可用作药物载体,但它们需要使用有机溶剂的复杂生产方法,在捕获药物方面效率低,并且难以大规模执行。固体脂质纳米颗粒(SLN)和纳米结构脂质载体(NLC)旨在解决其中的一些缺点。传统脂质体包含液晶态双层脂质,SLN包含固体脂质,NLC包括固体和液晶脂质的混合物。SLN和NLC的粒径在40到1000 nm之间变化。SLN和NLC表现出增强的物理稳定性,解决了基于脂质体的制剂的主要局限性之一。SLN和NLC还具有更高的装载能力和更高的货物生物利用度,无需使用有机溶剂即可轻松大规模生产,并且与其他LNP相比,灭菌过程中更为稳定。此外,分子在固态下的迁移率降低使SLN和NLC能够更精确地控制其药物有效载荷的释放。然而,在长期储存中,SLN的结晶会将药物成分排出到周围的介质中。通过在室温下将少量脂质液体引入SLN来设计NLC,可降低脂质核的结晶度。NLC结晶度的降低抑制了药物从基质中的排出,增强了纳米颗粒的载药能力和理化长期稳定性。
和纳米结构脂质载体(右)的示意图")
图4 固体脂质纳米颗粒(左)和纳米结构脂质载体(右)的示意图
SLN和NLC通常采用各种无溶剂方法生产,如高压均质、高速搅拌、超声、乳液/溶剂蒸发、相转化和溶剂注入。
临床批准的基于LNP的药物
50多年来,脂质体一直被认为是医学的有力工具。它们能够可控地将治疗药物封装和输送到体内的特定位置,用于治疗各种疾病。许多LNP药物制剂已被批准并用于医疗实践。LNP在药物递送中最大的单一应用是癌症治疗,因为LNP包封的抗肿瘤药物比游离药物具有更高的生物利用度和选择性。脂质纳米载体降低了抗癌药物对正常组织的毒性,增加了疏水药物的水溶性,延长了药物停留时间,提高了对药物释放的控制。

图5 批准的LNP药物及其针对的疾病
LNP还通过增强的渗透性和保留性(EPR)效应来提高癌症治疗的疗效。肿瘤中快速但有缺陷的血管生成导致血管具有较大的窗孔(>100 nm大小),LNP可以很容易地通过这些窗孔。因此,肿瘤血管对LNP的渗透性要强得多。当静脉内给药时,它们会在肿瘤中选择性积累。此外,肿瘤中功能失调的淋巴回流降低了LNP离开肿瘤的速度,从而提高了它们的保留。由于EPR效应,LNP在肿瘤中的积累允许纳米颗粒在肿瘤细胞附近选择性地释放抗肿瘤剂。
Doxil是最早批准的抗癌纳米制剂,也是最早批准的脂质体药物。该配方旨在改善蒽环类药物阿霉素的药代动力学和组织分布。阿霉素是一种有效的抗癌剂,但具有心脏毒性。Doxil 的EPR效应,使用空间稳定纳米颗粒延长人血浆中的循环时间,同时降低阿霉素的心脏毒性。它是作为静脉注射剂开发的,用于治疗晚期卵巢癌、多发性骨髓瘤和HIV相关的卡波西肉瘤。
核酸疗法是一类新兴药物,具有治疗各种疾病的潜力。然而,由于核酸是多价阴离子,具有高度亲水性,它们几乎不会被细胞吸收。同时,它们也很容易被血液中的核酸酶降解。因此,它们需要递送载体才能进入细胞并发挥效用。LNP载体是递送核酸药物的成功方法之一。核酸药物Patisiran(ONPATTRO)是一种在LNP中配制的siRNA,用于减少肝脏中转甲状腺素蛋白的形成,最近获得了FDA批准用于治疗遗传性转甲状腺素介导的淀粉样变。它是最早获批的siRNA药物,也是最早的LNP制剂核酸药物,标志着核酸治疗学发展的重要里程碑。
COVID-19 mRNA疫苗中的LNP
LNP成功使用于辉瑞/BioNTech和Moderna最近批准的两种COVID-19 mRNA疫苗的递送载体。这两种疫苗的开发速度无与伦比,在预防疾病方面显示出显著的有效性。疫苗将编码SARS-CoV-2刺突蛋白的mRNA递送到宿主细胞的细胞质中;mRNA被翻译成刺突蛋白,该蛋白充当抗原并导致对病毒的免疫反应的发展。
两种mRNA疫苗的脂质纳米颗粒的组成非常相似。两者都含有可电离脂质,在低pH下带正电荷(与RNA络合)和在生理pH下中性(减少潜在的毒性作用并促进有效载荷释放)。它们还含有聚乙二醇化脂质(PEG-脂质),以减少调理作用和吞噬细胞的清除,从而延长体循环时间。二硬酯酰磷脂酰胆碱(DSPC)和胆固醇有助于将货物装入LNP。辉瑞公司疫苗的阳离子脂质∶PEG-脂质∶胆固醇∶DSPC的摩尔比为46.3∶1.6∶42.7∶9.4,莫德纳疫苗的摩尔比为50∶1.5∶38.5∶10。这些纳米颗粒的直径为80~100 nm,每个脂质纳米颗粒含有约100个mRNA分子。
辉瑞专有的阳离子脂质——ALC-0315和Moderna专有的阳离子脂质——SM-102,两种脂质都是叔胺,在低pH值下质子化(因此带正电荷)。它们的烃链通过可生物降解的酯基团连接,可在mRNA递送后实现安全清除。mRNA疫苗中使用的阳离子脂质含有支链烃链,可优化非层状相的形成和mRNA递送效率。PEG-脂质都是PEG-2000偶联物。LNP在低pH(pH4.0)下制备,此时可电离脂质带正电,因此它可以很容易与mRNA形成复合物。微流控装置可将含有mRNA的液流与含有乙醇的脂质混合物液流混合。当快速混合时,这两种液流形成纳米颗粒,捕获带负电荷的mRNA。

图6 COVID-19 mRNA疫苗LNP的脂质成分结构
临床试验中基于LNP的mRNA疫苗和治疗方法
mRNA疫苗和疗法在预防和治疗疾病方面具有很大的前景。LNP启用的mRNA细胞内递送允许在宿主细胞内表达几乎任何所需的蛋白质。基于mRNA的疗法的一个重要特征是插入诱变的风险低。与DNA疗法不同,mRNA不需要细胞核的机制来执行其任务。由于mRNA不整合到宿主基因组中,因此降低了基于mRNA的疗法致癌和诱变的风险,从而提高了其安全性。最后,mRNA的制造比DNA的生产更容易标准化,并且提供了更好的可重复性。
mRNA疫苗因其效率高、开发周期短、成本低、生产潜力高而彻底改变了疫苗开发。如果没有LNP输送核酸技术的进步,就不可能快速开发mRNA疫苗。基于LNP的mRNA疫苗已进入针对多种传染病的临床试验,例如针对寨卡病毒,巨细胞病毒,结核病和流感。mRNA治疗性疫苗在针对黑色素瘤、卵巢癌、乳腺癌和其他实体瘤的癌症免疫治疗方面也具有巨大潜力。LNP载体对于将mRNA成功递送到免疫细胞的细胞质中至关重要,特别是负责触发所需免疫应答的抗原呈递免疫细胞。
免疫组织化学技术(Immunohistochemistry Technique,IHC)是在免疫学、生物化学和显微镜技术基础上发展起来的免疫细胞化学技术。它利用可见的生物标记物标记特异性抗体,通过抗原抗体反应和组化呈色反应对相应组织细胞抗原进行定位、定性或定量测定的一项免疫检测方法。该技术巧妙地将免疫反应的特异性、组化的可见性和分子生物学技术的灵敏度有机地结合起来,借助荧光显微镜及电子显微镜的显像和放大作用,在细胞、亚细胞水平检测多种化学组分(如蛋白质、多肽、多糖、酶、激素、病原体及受体等),为疾病的诊断、鉴别诊断及发病机制的研究提供了重要手段。
免疫组化技术根据标记物的不同可构成不同的免疫组化方法,如使用荧光素或荧光蛋白标记可构成荧光免疫组化技术、使用酶标可构成酶免疫组化技术、标记金或铁可构成免疫胶体金或胶体铁组化技术、使用生物素标记可构成亲和组化技术。
荧光免疫组化技术(fluorescence immunohistochemistry technique)是采用荧光素标记的已知抗体(抗原)作为探针,检测待测组织、细胞标本中的靶抗原(或抗体),通过抗原抗体特异性反应,形成带有荧光素的特异性抗原抗体复合物,在荧光显微镜下,这种复合物上的荧光素受激发光照射而发出各种颜色的荧光,据此可以分辨出抗原(或抗体)所在位置或性质,并可利用荧光定量技术计算其含量,实现定位、定性和定量测定的目的。蛋白质、核酸、多肽、酶、多糖、激素、受体及病原体等凡可作为抗原或半抗原的物质均可用荧光免疫组化技术检测。
酶免疫组化技术(enzyme immunohistochemistry technique)是在免疫荧光技术基础上发展起来的,其基本原理与荧光免疫组化技术相同,所不同的是酶免疫组化技术是用酶标记抗体,与免疫荧光技术同为免疫组化中最常用的方法。
显色反应:酶+底物=不溶性的色素
常用的酶是辣根过氧化物酶(HRP)、碱性磷酸酶(ALP)、葡萄糖样化酶(GOD)等,常用的供氢体有二氨基联苯胺(DAB)。
免疫胶体金技术是一种以胶体金为标志物的技术。这种特殊的金属颗粒是金的水溶胶形式,可以快速稳定地结合蛋白质,且对蛋白质的生物活性几乎没有影响。因此,胶体金是一种理想的材料,可以与一抗、二抗或其他一些可以结合IgG(如葡萄球菌A蛋白)的蛋白质结合,以定位切片组织或培养细胞中的某些抗原。由于胶体颗粒大小的差异及其较高的电子密度,免疫胶体金技术适用于免疫电子显微镜和光学显微镜下的单标记或多标记检测。
亲和组化技术(affinity histochemistry technique)是以一种物质对某种组织成分具有高度亲和力为基础建立的组化技术。一些具有双价或多价结合力的物质如植物凝集素(lectin)、生物素(avidin)和葡萄球菌A蛋白(SPA)等,对某种组织成分具有高亲和力及多级放大效应,能偶联抗体等大分子生物活性物质,并可与荧光素、酶、放射性核素、铁蛋白及胶体金等标记物有机结合,采用荧光显微镜、酶加底物的显色反应、放射自显影或电子显微镜等,在细胞和亚细胞水平进行对应亲和物质的定位、定性或定量分析。该技术具有高灵敏度、操作简单、省时等优点,尤其是具有多级放大效应,因而更有助于微量抗原(或抗体)在细胞和亚细胞水平的定位、定性或定量分析。
主要包括:
组织材料的处理对于免疫组织化学技术至关重要。在组织细胞材料准备的过程中,不仅要求保持组织细胞形态的完整性,保持化学成分和酶的活性,更要保持组织细胞成分的抗原性。
标本的来源主要有以下几种:
①活体组织各种实验动物和人体活检组织
②各种体液及穿刺液
③培养细胞
STEP 1:标本固定
将组织置于某些化学试剂中,使细胞内的物质尽量接近于其生活状态时的形态结构和位置称为固定,其基本原则是不损伤细胞形态、不干扰固定后抗体对抗原的识别和结合。
常用的固定方法有浸泡法、蒸汽法、注射灌注法、微波法。
STEP 2:制片
免疫组化最常用的制片方法是冷冻和石蜡切片。为了使抗原达到最大限度的保存,首选的制片方法是冷冻切片。其操作简便,可避免石蜡切片因固定、脱水、浸蜡等对抗原所造成的损失,适用于不稳定的抗原。研究形态学的主要制片方法是石蜡切片,它不但是观察组织细胞结构的理想方法,而且可用在陈旧石蜡包埋材料免疫组织化学的回顾性研究。石蜡切片的优点是切片薄而有连续性,可长期保存。但对抗原的保存不如冷冻切片。
STEP 3:抗原修复
在制片过程中,甲醛使得组织切片中的蛋白质发生凝固,在凝固过程中可引起蛋白质自身和不同的蛋白质之间通过甲基化桥使蛋白质发生交联,广泛的蛋白交联可导致蛋白质的三级或四级结构的折叠方向发生改变,同时抗原与其他分子间也会发生交联,致使抗原信号减弱或消失。因此,使组织抗原决定簇重新暴露,即抗原修复是免疫组织化学技术中的重要步骤。
非特异性背景染色原因与解决方法:
1)操作过程种漂洗不充分,可增加漂洗次数和漂洗时间;2)加试剂后未保湿导致切片变干,需在湿盒中反应避免切片变干;3)组织切片发生折叠;4)未阻断组织种含有过氧化物酶,可使用新鲜配置的3%双氧水封闭并延长孵育时间;5)未封闭组织内源性生物素,可使用阻断剂阻断内源性的生物素;6)组织抗原发生弥散;7)切片黏附剂过厚;8)组织蛋白封闭不充分。
染色强原因与解决方法:
1)一抗浓度过高或抗体孵育时间过长;2)孵育温度过高,应该在室温20~28℃孵育,或4℃过夜;3)生物素化二抗孵育时间过长;4)DAB显色时间过长或DAB浓度过高,应保证显色时间不超过5~10分钟。
染色弱原因与解决方法:
1)抗体浓度过低或孵育时间过短,可增加抗体浓度,且保证服务时间不少于60 min;2)室温过低,可适当延长孵育时间内;3)组织固定和包埋处理后抗原被破坏,应及时固定新鲜组织,防止自溶;4)蛋白过度封闭,可适当缩短封闭时间;5)复染或衬染太深。
染色假阴性原因与解决方法:
1)一抗与二抗不匹配;2)组织中可能无抗原,建议设立阳性对照实验组。
参考文献
[1]王兰兰,许化溪.临床免疫学检验(第5版)[M].北京:人民卫生出版社,2012
[2]吴秉铨,刘彦仿.免疫组织化学病理诊断[M].北京科学技术出版社,2007.
酶免疫组织化学技术(enzyme immunohistochemistry technique,EIT)是免疫组织化学中最常用的方法之一,是在一定的条件下,应用酶标记抗体(抗原)与组织或细胞标本中的抗原(抗体)发生特异性免疫反应,在酶的催化作用下催化底物产生显色反应,通过显微镜观察标本中抗原(抗体)的分布位置和性质,也可通过图像分析技术达到定量的目的。
酶免疫组织化学具有以下优点:①无需特殊显微镜设备;②定位准确,且对比度好;③酶标染色标本可长期保存;④可使用苏木素-伊红染料复染,与形态学相结合,便于观察结果;⑤显色反应的底物易分辨,电子密度大,便于光镜及电镜观察;⑥根据酶和底物的不同显示不同颜色,可进行双重或多重染色。尤其是非标记抗体酶法的灵敏度更优于荧光免疫技术。由于以上优点使酶免疫组织化学技术成为临床病理诊断、肿瘤性质判断及预后观察的重要手段。
酶标记抗体
借助交联剂共价键将酶直接连接在抗体上形成酶标抗体,酶标抗体与靶抗原反应后,通过酶对底物的特异性催化作用,生成不溶性有色产物。常用的方法有直接法和间接法。
①直接法:将酶直接标记在特异性抗体上,与组织细胞内相应抗原进行特异性反应,形成抗原-抗体-酶复合物,最后用酶底物显色。直接法操作简便但灵敏度低,此外需要依据不同抗原制备相应的酶标抗体。
②间接法:将酶标记在第二抗体上形成酶标二抗,先将第一抗体(特异性抗体)与相应的组织抗原结合,形成抗原抗体复合物,再用第二抗体(酶标二抗)与复合物中的特异性抗体结合,形成抗原-抗体-酶标二抗复合物,最后用底物显色剂显色。间接法检测灵敏度高,制备一种酶标二抗可用于检测多种抗原或抗体,但特异性不如直接法,可能会出现假阳的情况。
非酶标记抗体
非酶标记抗体技术使用酶免疫动物,制备效价高、特异性强的抗酶抗体,通过免疫学反应使得抗酶抗体与组织抗原结合。该方法避免了酶标记时对抗体的损伤,同时也提高了方法的灵敏度。它有以下几种技术类型:
①酶桥法
抗酶抗体作为第三抗体,通过桥联抗体(第二抗体),将特异性识别组织抗原的第一抗体与第三抗体连接起来,形成酶联的抗原-抗体复合物,加底物显色。酶桥法较酶标法的灵敏度有所提高,但操作更为复杂。在酶桥法中,如果抗酶抗体与酶结合弱,在操作中酶常被冲洗掉;如果酶标记在非特异性抗体上就会存在背景着色问题;同时抗酶抗体的非特异性成分也能竞争结合桥联抗体的结合位点,影响方法的灵敏度。


图 1 PAP复合物

与酶桥法相比,PAP复合物结构稳定,避免了酶桥法中酶标记易脱落的弊端;灵敏度高,较桥酶法灵敏20倍;PAP是一种复合物,无游离免疫球蛋白存在,不易引起非特异性染色。
(三)双桥PAP法
双桥PAP法的基本原理是通过两次连接桥联抗体和PAP复合物,使得双桥可结合更多的PAP复合物于抗原分子,以增强灵敏度。这种使用桥联抗体放大的方法,使桥联抗体与PAP复合物中抗酶抗体的未饱和Fc段结合,或桥联抗体与特异性第一抗体尚未饱和的Fc段结合,从而达到对抗原的明显放大效果,对于组织细胞微量抗原的检测有实用价值。
参考文献
[1]王兰兰,许化溪.临床免疫学检验(第5版)[M].北京:人民卫生出版社,2012
[2]吴秉铨,刘彦仿.免疫组织化学病理诊断[M].北京科学技术出版社,2007.
单克隆抗体已成为治疗和诊断人类疾病的有效工具。非人源抗体会诱导人类免疫反应,使用非人源抗体产生的中和反应会限制抗体药物在治疗人类疾病中的应用。为了克服这个问题,抗体人源化技术应运而生。到目前为止,研究人员已经开发了各种方法用于非人源抗体的人源化,并提高其亲和力、特异性等其他特性。本文回顾了各类抗体人源化技术的相应进展,并针对这些方法列举了一些重要研究。
非人源抗体人源化的常用方法是互补决定区(CDR)移植,即将非人源抗体的CDR区移植到人源抗体框架区上。通常,会选择与非人源抗体框架区同源性最高的人源抗体框架区作为CDR移植的受体。这种方法最主要问题是,改造后的抗体与特定靶标结合的亲和力会降低乃至丧失。将鼠源抗体的CDR环直接移植到人源抗体框架上仅在少数情况下不会影响抗体亲和力,而大多情况下抗体亲和力会显著降低。鼠源抗体框架区的一些残基已被证明会影响CDR环的构象以及抗体的亲和力,我们称其为游标区残基。这些残基位于靠近CDR区的β折叠。因此,在选择所需的人源抗体框架区后,需要对这些残基进行回复突变,使其保留在人源化抗体中。例如,赫赛汀(曲妥珠单抗)是FDA批准的抗HER2抗体,是通过将鼠源抗体4-5D的CDR环移植到人IgG框架上并保留游标区氨基酸而产生。Makab及其同事为了了解游标区残基对CDR构象的影响进行了一项实验,他们通过直接移植CDR改造了识别表皮生长因子受体(EGFR)鼠源抗体的人源化版本,发现其亲和力损失为1/40。亲和力的降低是由于与结合相关的负焓变化显著减少。人源化后,无配体抗体片段的晶体结构没有明显的构象变化,人源化抗体的CDR环结构与鼠源抗体的环结构相同。为了确定哪类游标区残基是影响抗体亲和力的主要因素,他们表达了几种人源化抗体的回复突变版本,其中一些游标区残基被反向突变为亲本鼠抗中的残基。几个单突变和一个双突变增加了负焓变化。然而,回复突变的个数的增加会导致负焓变化减少。这些结果表明,游标区残基对抗原结合有焓变贡献,但同时必须仔细考虑和优化鼠源抗体人源化时构象熵变化的调节。
除可变区氨基酸残基的回复突变外,可变区外氨基酸残基的突变也已被用于赋予人源化抗体新的特性。
人类胚系基因可作为鼠源抗体人源化框架区的替代来源。与来源于IgG的框架区相比,胚系基因具有较少的克隆内体细胞超突变。因此,人们认为利用胚系框架的人源化抗体比IgG框架的人源化抗体表现出更低的免疫原性。有研究使用该方法对鼠抗WO-2进行人源化。WO-2是一种针对与阿尔茨海默病相关的Aβ肽产生的单克隆抗体。通过保留鼠抗游标区残基以保持亲和力,研究人源构建了该抗体的三种人源化形式,包括单链抗体、Fab片段和含有人类恒定区域的嵌合Fab片段。他们发现,这种抗体的单链和Fab片段形式与Aβ肽结合,其亲和力比重组嵌合Fab片段低约两倍,比木瓜蛋白酶裂解的亲本Fab片段低五到六倍。尽管鼠WO-2的人源化导致亲和力降低,但问题不大,人源化后抗体对Aβ仍具有高亲和力(在纳摩尔范围内)。
尽管胚系基因的免疫原性可能较低,但事实上IgG的衍生框架有时更有利。复数研究反应,人源化抗体Fc区的变化会影响抗体活性与亲和力。如有研究证实,将鼠抗可变区融合到IgG恒定区构建的嵌合抗体对黄热病感染的预防和治疗有效,而具有IgM恒定区的嵌合抗体则不然。
抗体表面重塑是非人源抗体人源化的另一种策略。表面重塑是指对非人源抗体的表面氨基酸残基进行抗体人源化改造。该策略的原则是确定鼠源抗体表面残基的位置,在维持抗体活性兼顾减少抗体免疫原性的原则下,选用与人源抗体表面残基相似的氨基酸进行替换。通过这种方法人源化的抗体通常表现出稳定性和亲和力的变化很小。
如人源化抗体h82D6A3,其可变区含有用于CDR环构象的必要鼠源抗体框架残基,框架区的10个表面残基中与亲本鼠抗体的可变区不同。与亲本鼠抗体82D6A3相比,它的亲和力略有下降。小鼠抗肿瘤坏死因子-α(TNF-α)抗体m357也通过该方法人源化。该抗体在其框架区中含有17个非保守表面残基,其中6个被突变为PPS4中的残基(PPS4是一种具有与m357可变区同源的人源抗体)。其他表面残基已在人源化抗体中保留,因为研究者认为它们是支持CDR环构象所必需的。
上述的CDR移植通常会选择与非人源抗体框架区同源性最高的人源抗体框架区作为CDR移植的受体。Hwang及其同事首次设计了一种基于CDR区域同源性的抗体人源化新方法。该方法不使用框架区的同源性来选择人源化抗体框架,关键的鼠源残基也不进行回复突变。使用这种方法可以减少被识别为外源物质的可能性。比起基于框架区同源性的CDR移植,通过该方法改造的抗体亲和力维持更好。
通过CDR移植获得的人源化抗体仍可能在患者中引发免疫性抗独特型(anti-Id)反应。为了最大限度地减少抗V区免疫反应,可以通过仅将CDR序列中抗原结合活性所必需的特异性决定残基(SDR)移植到人源抗体框架区上来实现抗体人源化。SDR移植的方法更进一步地提升了抗体人源化程度,并尽可能地减少了鼠源CDR中效应T细胞表位的数量,从而将抗体可变区潜在的免疫原性风险做到最小化。
链替换抗体库技术
链替换抗体库技术(或定向筛选)是利用噬菌体展示的方法,将鼠源抗体重轻链V区结构域分别顺序或平行地替换为人源化的。该方法为人源化提供了一个强大的工具,可以最大限度地减少人体的免疫原性。值得注意的是,即使是通过噬菌体展示技术产生人源化抗体,哪怕是全人源抗体,也可能显示出一定程度的免疫原性。
基于human string content优化
该策略基于一种与免疫学相关的抗体人源化指标,称为human string content(HSC)。HSC的得分在具有不同人类胚系序列框架和CDR区的小鼠氨基酸域中显著不同。目标序列的人源化是通过最大化该参数,而不是使用广泛的一致性度量生成多种多样的人源化改造抗体。所得可变区的免疫原性较低,并显示出与通过标准CDR移植方法人源化的结合亲和力相当的增强。Hammond及其同事使用HSC方法将嵌合抗CD30抗体cAC10人源化。新型Xmab2513抗体对FcgRIIIA受体和CD30抗原的结合亲和力显著提高(比亲代抗体高4倍),并保留了cAC10和5F11抗体所表现出的强大抗增殖作用。
到目前为止,已经开发了多种抗体人源化方法,每种方法各有优劣,可以根据具体情况,选择其一。抗体的所有特性,包括免疫原性、亲和力和特异性,都可以通过改变CDR或框架区中的一些氨基酸残基来实现。考虑到人源化抗体在人类疾病治疗中的应用范围广泛,应用日益增多,未来该领域必将取得更多发展。
参考文献
[1]Safdari Y, Farajnia S, Asgharzadeh M, et al. Antibody humanization methods-a review and update[J]. Biotechnol Genet Eng Rev. 2013(29):175-186. doi:10.1080/02648725.2013.801235
[2]Ahmadzadeh V, Farajnia S, Feizi MA, et al. Antibody humanization methods for development of therapeutic applications[J]. Monoclonal antibodies in immunodiagnosis and immunotherapy. 2014;33(2):67-73. doi:10.1089/mab.2013.0080
免疫分析技术(IA)是以抗原和抗体的特异性、可逆性结合反应为基础的分析技术,随后相继发展起来的酶免疫分析法(EIA)、荧光免疫分析法(FIA)和化学发光免疫分析法(CLIA)等免疫学分析方法,凭借其高灵敏性、高特异性、高通量、迅速及价格低廉的特点,在药物残留分析、环境污染物检测等领域得到了广泛应用。
大多数小分子药物、毒素、环境污染物分子质量小于1000 u,属于仅有免疫反应原性而无免疫原性的半抗原。目前制备小分子半抗原抗体的常规方法为:选择具有毒理学意义的代谢产物或原形药物作为待测物。设计合成保留待测物分子结构特征并带有活性基团的半抗原,通过共价键使半抗原与大分子质量蛋白质载体偶联,制备人工免疫原,经动物免疫制备针对半抗原的特异性抗体。但对于分子质量较小的半抗原,尤其是分子质量小于300 u的半抗原,常常难以获得高亲和力抗体。这一问题可通过尝试新型检测模式和级联放大系统等方式进行优化,但与此同时,对检测条件和检测设备也提出了更高的要求。因此,现阶段研发方向已转移到优化半抗原设计与合成上来。
半抗原设计的目的是使半抗原刺激机体产生特异性免疫应答,并获得对待测物分子具有高亲和力的抗体。半抗原的设计中须遵循以下原则:
1.人工免疫原中的半抗原应在分子结构立体化学和电子分布上与待测物分子尽可能相似;
2.半抗原结构中的连接臂应不易于诱导产生“臂抗体”,最好使用一定长度的碳链;
3.半抗原分子应具有便于与蛋白载体偶联的活性基团(如:NH2COOH、OH、SH等),且活性基团的存在对待测物分子的电子分布应没有影响;
4.半抗原与蛋白偶联后仍应保留待测物分子的基本结构。
因此,半抗原设计的关键在于尽可能保留原半抗原的特征结构,并在适当的位置引入合适的连接臂和与蛋白载体偶联的活性基团。
通常半抗原分子有多个可供选择的修饰位点,但选择不同的修饰位点后期暴露出的抗原决定簇不同,这会导致产生的抗体在效价、亲和力和特异性上都存在差异。
当修饰位点与半抗原分子的特征结构(如-COOH,-NH,-OH,环或杂环等)距离较近时,不利于半抗原分子特征结构的暴露,使得动物的免疫系统对半抗原分子特征结构的识别难度加大。因此半抗原分子中的修饰位点应远离半抗原的特征结构部分和官能团,使半抗原的特征分子结构部分得以充分暴露。另外,在半抗原分子的不同修饰位点进行化学修饰时,可能会导致半抗原分子中电子分布的改变,从而使半抗原分子的电化学性质发生改变,导致免疫原免疫活性的改变。
通常一个半抗原分子有多个不同的修饰位点时,可利用不同的修饰位点合成出几种半抗原修饰物,选择半抗原分子结构保持最完整的修饰物与载体相连制备人工免疫原;同时可以选择半抗原分子结构保持得不够完整的修饰物与载体相连制备人工免疫原。当半抗原修饰物的分子结构与原有半抗原的结构差异性越大,抗体的亲和力越弱,越有可能建立起具有高灵敏度的测定小分子化合物的间接竞争免疫分析方法。
半抗原特征分子结构与载体之间的连接部分为连接臂。引入连接臂的目的主要是为了在人工免疫原表面突显出半抗原的特征结构。连接臂的选择应该遵循以下基本原则:
1.连接臂的连接应尽量避免在目标半抗原的官能团处或靠近官能团处,最好位于重要的特征性官能团的远端。结构类似的一类化合物连接臂应当在它们具有相同结构或类似结构的位置连接以最大限度的使分子的特征部位暴露;
2.连接臂长度要适宜。连接臂太短,则载体的空间位阻影响半抗原特征结构的暴露,且半抗原结构容易受到载体局部化学环境的影响发生变化;连接臂太长,则可能因某些极性连接(氢键)或非极性连接(疏水作用)使半抗原发生“折叠”。通常认为3~6个碳原子直链是最佳的长度;
3.避免连接臂中含较强的决定簇(如芳环、共双键或杂环等),通常以含末端活性基团的链环烃为宜,以减少所产生的抗体对连接臂的过度识别。
活性基团的引入有两种方式:第一种是利用待测物上已有的活性基团通过双功能试剂引入连接臂和活性基团,使之与载体蛋白偶联。这种方案的优点是比较容易实施,但对于一些待测物,若这些活性基团是其特征结构,或偶联后改变了分子的电子分布,则会影响抗体对待测物的识别。第二种方式是直接合成待测物的带有(CH)nCOOH、(CH)nNH等结构的衍生物,这种方案有利于保护待测物的特征结构和分子的电子分布不受影响,但合成上有时比较困难,往往需要多步反应才能实现。
半抗原偶联载体不仅可增加半抗原的相对分子质量,或仅起到运载作用而且可依靠本身的结构特异性和免疫原性,诱导机体产生免疫应答反应继而诱导对半抗原分子的识别这种现象称为载体效应。常见载体一般为蛋白质,如球蛋白片段、牛血清白蛋白(BSA)、鸡卵清蛋白(OVA)、钥孔血蓝蛋白(KLH)、兔血清白蛋白(RSA)、人血清白蛋白(HAS)、甲状腺球蛋白、纤维蛋白原或兔和鸡的丙种球蛋白等。其中,最常用的载体是牛血清白蛋白(BSA)。BSA物化性质稳定、不易变性且赖氨酸含量高,分子内有很多自由的氨基,在不同的pH值和离子强度下均有较大的溶解度。钥孔血蓝蛋白(KLH)因其与脊椎动物免疫系统具有很好的异源性而被认为是优先选择的载体。
半抗原修饰物与载体偶联的常用方法有化学偶联法、化学生物学方法和免疫学标记法。其中以化学偶联法最常用。一般可根据半抗原修饰物所含活基团的不同,选择不同的方式与载体蛋白进行偶联。如半抗原修饰物含羧基、则可通过碳化二亚胺法、混合酸酐法、活性酯法与载体蛋白进行偶联;半抗原修饰物含氨基,则可通过戊二醛法、二异氰酸酯法、卤代硝基苯法、亚胺酸酯法等与载体蛋白进行偶联;半抗原修饰物含羟基,则可通过琥珀酸酐法、偶氮苯甲酸法、一氯醋酸钠法等与载体蛋白进行偶联;若半抗原修饰物含疏基,可通过SAMSA反应与载体蛋白质通过二硫键交联;若半抗原修饰物含醛基或酮基,可通过O-(羧甲基)羟胺法和对肼基苯甲酸法合成带有羧基的中间体,再通过羧基与蛋白质的氨基结合。
德泰生物小分子抗体服务基于兔单抗SingleB®快速发现技术,提供从小分子抗原合成&偶联到动物免疫、单B细胞筛选、抗体测序与制备纯化的一站式服务,可作为ADC抗体药物PK检验中小分子载荷的捕获抗体,检测与小分子药物偶联的抗体、小分子药物可能转移到的血清白蛋白或游离小分子。
自然界中抗原结构复杂、种类繁多,并且含有多种表位。面对抗原的刺激,机体可以产生抗原表位特异性抗体,以及针对同一表位的不同类型抗体。
Ig基因重排
人的Ig重链基因由编码可变区的V基因片段(VH)、D基因片段(DH)、J基因片段(JH)以及编码恒定区的C基因片段组成,VH、DH和JH的基因片段数分别为45、23和6个。Κ或λ轻链基因可变区由V、J基因片段组成,Vκ和Jκ基因片段数分别为40和5个,Vλ和Jλ基因片段数分别为30和4个;重组C基因片段有9个,按照5’-Cμ-Cδ-Cγ3-Cγ1-Cα1-Cγ2-Cγ4-Cε-Cα2-3’的顺序排列。Cκ基因片段数只有1个,Cλ基因片段数有4个,按照5’-Cγ1-Cγ2-Cγ3-Cγ7-3’的顺序排列。Ig胚系基因以被分隔开的基因片段成簇存在,通过重组酶的作用,从众多V(D)J基因片段中各选择一个V片段、D片段(轻链无)、J片段,形成V-D-J(重链)或V-J(轻链)连接,再与C基因片段连接,编码形成完整的Ig多肽链,进一步加工、组装成有功能的BCR。在B细胞分化发育过程中,BCR基因片段发生重新排列和组合,产生数量巨大、能识别特异性抗原的BCR。
人类Ig基因片段可以产生多种V区基因片段组合,理论上仅重链V区的排列就可达45(VH)×23(VD)×6(VJ)=6210之多,轻重链组合多样性可达1.9×106。因V区基因片段使用频率及轻重链配对成功概率不同,实际组合数量要比理论上少一些,但仍具有丰富的多样性。此外,Ig基因片段还可以通过密码子错位、框架移位以及N序列插入等形式产生新的序列,从而增加BCR多样性。
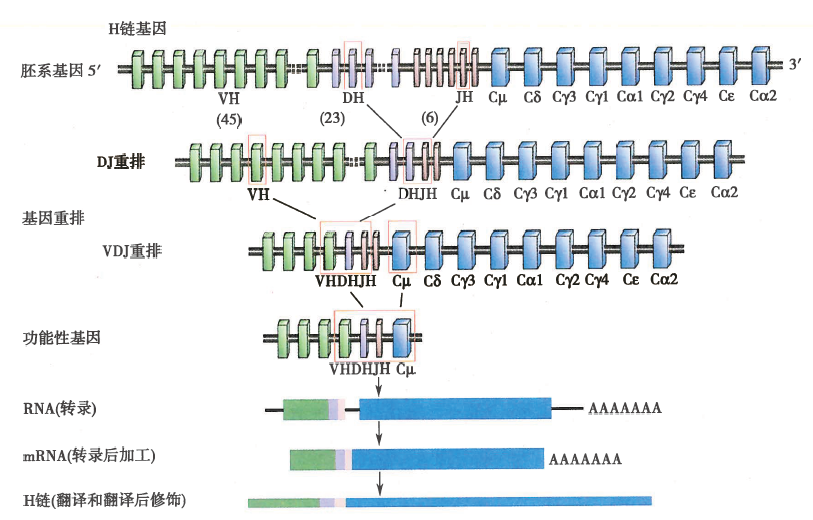
图1 免疫球蛋白重链基因重排及表达示意图
注:重链胚系基因经过重排先形成D-J连接,然后形成V-DJ连接,编码功能性V区基因。
抗体类别转换
2020年Cell Reports上发表了一篇题为“Antibody Isotype Switching as a Mechanism to Counter HIV Neutralization Escape”的文章。研究者发现在HIV感染的不同时段,体内的抗体同种型差异明显,同种型抗体是通过抗体类别转换重组(CSR)产生的,CSR是一种B细胞特异性DNA重排反应,且不同同种型的抗体对病毒具有不同的中和作用。其中IgG3和IgA1中和活性较好,IgG3类抗体能够通过抗体依赖的细胞毒作用(ADCC)发挥抗病毒作用。为了产生最佳的免疫反应,成熟B淋巴细胞通过将表达Ig重链恒定区(CH)的基因从Cμ交换到另一个下游CH基因,将IgM抗体转换为具有相同抗原特异性、不同效应功能的二级同种型(IgG、IgA 或IgE)抗体。
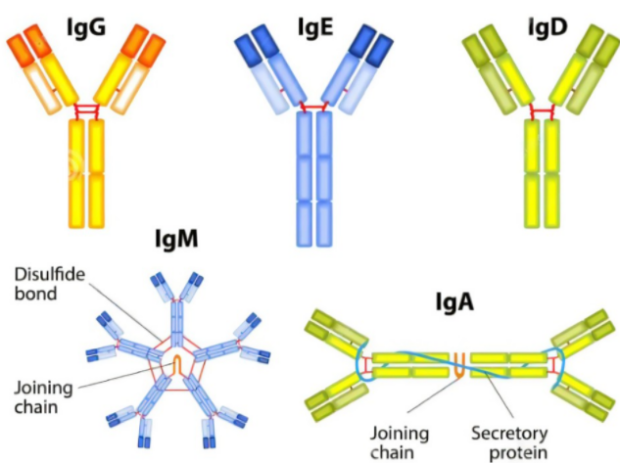
图2 抗体类别示意图
体细胞超突变(SHM)是另一种B细胞特异性反应,发生在CSR之后。一般体细胞基因突变率极低(1/1010-1/107),而在非常有限的B细胞染色体BCR V区基因片段中却能产生相当高频率的突变。在生发中心的活化B细胞每分裂一次,BCR的V区基因就会产生大约1/103碱基对突变。当遇到抗原时,抗原激活的成熟B细胞通过激活诱导胞嘧啶脱氨酶(AID)的活性启动SHM。
CH基因组转录过程中产生的转录泡、负超螺旋结构以及R-环产生的单链DNA招募激活诱导胞啶脱氨酶(AID)。AID将单链DNA S区上的胞苷(C)残基脱氨产生尿苷(U)。脱氨基的DNA被碱基切除、错配修复处理,发生双链DNA断裂(DSB)。双链DNA断裂使细胞内双链DNA损伤应答系统激活,最终非同源DNA末端连接系统将两个S区之间的 DNA 作为染色体外环丢失,完成DSB连接。
B细胞在外周淋巴器官生发中心接受抗原刺激后快速增殖,具有抗原亲和力低的B细胞通过在Ig可变区外显子部分引入点突变,对Ig基因进行修饰,以产生更高亲和力的抗体。
体细胞超突变可能会增加B细胞受体(BCR)对特定抗原的亲和力,当然也可能会导致BCR对抗原识别的丧失或产生自我反应性受体。大多数BCR亲和力降低甚至不表达BCR的突变B细胞克隆不能结合FDC(滤泡树突状细胞)表面的抗原,因无法将抗原呈递辅助性T淋巴细胞获取第二信号而凋亡;少部分突变B细胞克隆的BCR亲和力提高,表达抗凋亡蛋白而继续存活,从而实现了抗体亲和力的筛选。
B淋巴细胞由鸟类的法氏囊(bursa of Fabricius)或哺乳动物的骨髓(bone marrow)中的淋巴样祖细胞发育分化而来,因此称为B细胞。B细胞是免疫系统的重要组成部分,通过产生抗体发挥特异性体液免疫功能,同时也是一种抗原提呈细胞,参与免疫调节。

图1 B细胞发育主要阶段
B细胞发育过程根据分化发生的场所可以分为两个部分:
BCR
BCR是表达于B细胞表面的免疫球蛋白,即膜型免疫球蛋白(mIg)。B细胞通过BCR识别抗原,接收抗原刺激并进行体液免疫应答。B细胞分化发育过程中,BCR基因片段发生重新排列组合,从而产生数量巨大、能识别特异性抗原的BCR(VDJ重排原理)。
B细胞在骨髓中的分化过程中不受外来抗原的影响,也可以称为B细胞分化的非抗原依赖期。此过程中主要经历祖B细胞(pro-B cell)、前B细胞(pre-B cell)、未成熟B细胞(immature B cell)3个阶段,并伴随着两个主要事件——一是B细胞受体(BCR)的表达,二是B细胞自身免疫耐受的形成。
免疫细胞作为造血系统的重要组成部分之一,与其他造血细胞一样,来源于骨髓中的造血干细胞(HSC),骨髓中的血细胞根据其不同的功能可以大致分为淋巴系和髓系。HSC首先分化为淋巴系共同祖细胞(CLP)或髓系共同祖细胞(CMP),CLP随后根据不同细胞因子和细胞内转录因子的调控而继续分化。CLP一旦被决定分化为B细胞系,它们首先会分化为祖B细胞。祖B细胞是自我更新能力有限的祖先细胞,可分为early pro-B cell和late pro-B cell。在early pro-B cell阶段发生重链D-J重排,在late pro-B cell阶段发生重链的V-D-J重排。同时表达B系特异标志,如Thy-1+、Tdt+、B200+、mb-1+等分子。但整个pro-B cell阶段B细胞表面没有mIgM的表达。前B细胞阶段始于pre-BCR的表达。pre-BCR由μ链和替代轻链(包括与轻链V区和C区同源的VpreB和λ5两种蛋白)组成。pre-BCR信号引起B细胞克隆扩增,并能抑制H链VDJ进一步重排,保证等位基因排斥。pre-B cell也可分为两阶段,large pre-B和small pre-B,前述行为发生在large pre-B阶段,而在small pre-B阶段细胞停止增殖进而轻链开始进行V-J重排。
前B细胞在骨髓中发育至未成熟B细胞后,其表面仅表达完成完整的mIgM,此时的mIgM若与骨髓中的自身抗原结合,即会导致细胞凋亡,形成克隆清除,一些识别自身抗原的未成熟B细胞可以通过受体编辑改变其BCR的特异性。在某些情况下,未成熟B细胞与自身抗原的结合可引起mIgM表达的下调,这类细胞虽然可以进入外周免疫器官,但对抗原刺激不产生应答,称谓失能。在骨髓中发育的未成熟B细胞通过上述克隆清除、受体编辑和失能等机制形成了对自身抗原的中枢免疫耐受,成熟的B细胞到达外周淋巴组织后仅被外来抗原激活,产生B细胞适应性免疫应答。

图2 中枢免疫器官中的两大主要事件
当自身免疫耐受形成后,未成熟B细胞离开骨髓,迁移前往外周淋巴器官。在脾脏内,未成熟B细胞继续分化发育,形成过渡B细胞,主要包括不表达CD21 IgMhi IgDlo的过渡型T1 B细胞(Transitional B T1)和表达CD21 IgMhi IgDhi的过渡型T2 B细胞(Transitional B T2)。
过渡型T2 B细胞进入血液和次级淋巴器官,分化为边缘区B细胞(marginal zone B)或滤泡B细胞(follicular B)。FO B细胞是一类在次级淋巴器官里不断循环的长寿成熟B细胞,而MZ B细胞是在脾脏特定区域不参与循环的B细胞。MZ B细胞分子表面标记为CD21hi CD23–IgM+ IgDlo,FO B细胞分子表面标记为CD21lo CD23+IgMlo IgD+。

图3 淋巴结与生发中心
MZ B细胞和FO B细胞不仅在脾脏中所处位置不同,其在抗原特异性免疫应答中所起的作用也不同。多数抗原(TD抗原)仅能在有T细胞辅助的条件下诱导B细胞产生抗体,FO B细胞被认为参与由TD抗原诱导的抗体分泌过程。当遇TD抗原激活后,FO B细胞迁移到滤泡外区域或形成生发中心。生发中心是T细胞依赖性抗体应答过程中于外周淋巴组织内形成的一个特殊结构,可分为暗区(dark zone)和明区(light zone)。暗区密集分布着快速增殖的B细胞,即生发中心母细胞(CBs),增殖的B细胞发生体细胞高频突变,从而增加抗体亲和力,确保高亲和力的抗体可以形成;明区稀疏分布着由生发中心母细胞分化而来的生发中心细胞(CCs),以及帮助促进亲和力选择的滤泡树突细胞及滤泡辅助T细胞,在这里细胞经历抗体同型转换和亲和力成熟,从而筛选出高亲和力的B细胞。高亲和B细胞接收来自周围环境存活、增殖和分化的信号,最终分化成为记忆B细胞(memory B cell)或浆细胞(plasma B cell)。

图4 B细胞在外周淋巴器官的分化发育






南京德泰生物工程有限公司 Nanjing Detai Bioengineering Co.,Ltd. ©2026 All Rights Reserved
 苏公网安备32011202001300
苏公网安备32011202001300